黄铁矿(100)表面性质的密度泛函理论计算及其对浮选的影响
2011-11-08李玉琼陈建华
李玉琼,陈建华,陈 晔,郭 进
(1. 广西大学 化学化工学院,南宁 530004;2. 广西大学 资源与冶金学院,南宁 530004;3. 广西大学 物理科学与工程技术学院,南宁 530004)
黄铁矿(100)表面性质的密度泛函理论计算及其对浮选的影响
李玉琼1,陈建华2,3,陈 晔2,郭 进3
(1. 广西大学 化学化工学院,南宁 530004;2. 广西大学 资源与冶金学院,南宁 530004;3. 广西大学 物理科学与工程技术学院,南宁 530004)
采用密度泛函理论(DFT)平面波赝势方法计算理想黄铁矿(100)表面的结构弛豫、原子的Mulliken布居以及电子结构,并解释黄铁矿体相中电荷分布异常的原因。从浮选角度分析表面结构和性质对黄铁矿浮选行为的影响。结果表明:黄铁矿(100)表面弛豫较小,表面Fe-S相互作用相对于体相增强;表面5配位的铁原子具有较高的活性;表面层铁硫原子的能隙降低;表面层的导电性强于体相的,表面的电化学活性增强。
黄铁矿表面;密度泛函理论计算;Mulliken布居;电子结构;浮选
黄铁矿广泛存在于各种矿床中,经常与有色金属(如铜、铅、锌、锑和钼等)硫化矿共存,在采用泡沫浮选法回收这些金属矿物时,都会碰到与黄铁矿浮选分离的问题。另外,在煤矿中也经常存在黄铁矿,造成煤中含硫量超标,在煤的选矿中,通常采用浮选法脱除黄铁矿。矿物的浮选主要是通过表面与药剂发生相互作用,因此,研究黄铁矿表面的性质对于了解黄铁矿的浮选行为和药剂作用机理具有非常重要的意义。
目前,已有许多针对黄铁矿表面的实验测试研究[1−4],而黄铁矿的计算模拟可以在理想环境下清晰而直接地观察到原子层次的信息,可以帮助人们进一步了解黄铁矿表面性质。HUNG等[5]采用平面波赝势和高斯基组方法计算黄铁矿(100)面的结构驰豫和电子结构;von OERTZEN等[6]采用ab从头算量子化学模型预测黄铁矿(100)表面和体相之间的差异;CAI和PHILPOTT[7]采用ab从头算赝势方法计算黄铁矿(100)的表面态和表面能。黄铁矿的表面结构及性质对黄铁矿浮选具有重要意义,目前,大多数研究主要是集中在对黄铁矿表面性质的理论分析方面,而对于黄铁矿表面性质与其可浮性的关系则很少见到报道。
已经有较多的采用密度泛函理论方法(DFT)针对黄铁矿体相的研究结果发表[8−12],李玉琼等[13]研究了黄铁矿体相。由这些方法计算出来的黄铁矿体相性质,如晶格常数等,与实验值符合得非常好,但是仍有一些问题没有得到很好解决。如黄铁矿体相的铁原子和硫原子的Mulliken电荷布居计算问题,目前,国内外计算模拟的结果除了文献[14]外,其它报道的结果都是铁带负电硫带正电荷[6,15],而作者也没有对这样的结果作出具体的解释。王淀佐等[16]则认为是由于铁的3d轨道与硫形成配位键,得到较多的硫原子对的电子的缘故,但这仍与电负性理论相矛盾(硫的电负性大于铁),如果黄铁矿的铁带负电,这将无法解释浮选过程中药剂与黄铁矿中铁离子的作用,而且实际上黄铁矿中铁的价态是正的。
自然界中最常见的黄铁矿为立方晶体,(100)面是其最主要的解理面,解理过程中可能会有S—S键断裂,Fe—S键断裂,或者两者都断裂,留下不同的表面类。本文作者的计算采用的是沿Fe—S断裂、表面留下完整的S二聚体(S2)的理想(100)表面,并对其进行表面结构弛豫、Mulliken布居及电子结构计算,讨论表面与体相之间的差异,并根据表面电子结构和性质,研究黄铁矿与黄药和氧分子作用的电化学模型和机理。
1 计算方法
采用CASTEP软件[17−18]在密度泛函理论框架[19−22]下对黄铁矿(100)表面电子态密度、能带结构和布居进行第一性原理平面波赝势计算。表面模型从优化过的体相切出来,进行结构优化后再进行性质计算。在前面对体相进行计算的基础上[13],本研究中对表面进行优化计算的条件设为:采用(1×1)表面单胞模型;采用GGA下的PW91交换关联泛函;采用270 eV平面波截断能和4×4×1的Monkhorst-Pack k点取样密度[23−24];采用超软赝势[25]来描述价电子和核的相互作用。几何优化的收敛标准为原子位移的收敛阈值为0.000 2 nm,原子间作用力的收敛阈值为0.8 eV·nm,原子间的内应力收敛阈值为0.1 GPa,最大能量改变的收敛阈值为2.0×10−5eV·atom−1;自洽迭代收敛精度为2.0×10−6eV·atom−1;原子的赝势计算选取的价电子分别为S 3s23p4和Fe 3d64s2。对性质的计算采用与几何优化相同的参数,计算态密度的时候采用的smearing值为0.1 eV。表面计算的时候对所有原子都进行了驰豫。对不同原子层厚及真空层厚度进行测试,以确保近中心层的原子是似体相的,即可忽略它们的弛豫。所有的计算都采用自旋极化,并在倒易空间中进行。
2 结果与讨论
2.1 表面结构的弛豫
表1和2所列分别为不同原子层数及不同真空层厚度对表面能影响的结果。从表1可以看出,表面含有15层原子后,表面能的变化已经很小。从表2的结果可看出,真空层厚度为1.0 nm的表面的表面能最低。因此,15层原子及1 nm的真空层厚度的表面结构能够给出较满意的收敛结果。黄铁矿单胞及(100)表面单胞模型显示在图1中。
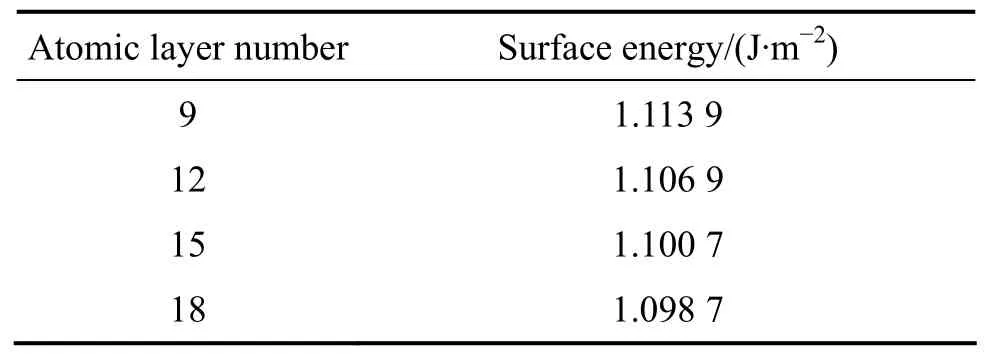
表1 不同原子层数的表面能Tabel 1 Energy of surface containing different atomic layer numbers

表2 真空层厚度对表面能的影响Tabel 2 Effect of vacuum layer thickness on surface energy

图1 体相黄铁矿单胞(a)及黄铁矿(100)表面单胞(b)Fig.1 Bulk pyrite unit cell (a) and pyrite (100)surface unit cell (b)
黄铁矿(100)表面结构和离子位移矢量如图1所示。表3所列为弛豫后表面几层原子的配位数及位移。体相黄铁矿的硫的配位数为4,铁的配位数为6,理想的黄铁矿(100)表面分布有3重配位的硫和5重配位数的铁。黄铁矿表面解理造成表面原子配位数降低,表面原子缺少周围原子的束缚因而产生了不同程度的弛豫。结合图1和表3可知,表面第一层S原子向表面内部弛豫;最明显的弛豫是第二层的表面Fe原子,向内部弛豫了大约0.01 nm;第三层中的S原子向表面弛豫。计算结果表明:黄铁矿理想(100)面经历的弛豫非常小,没有产生明显的表面重构作用,这与ROSSO等[2]及CHATURVEDI等[3]的实验测试结果一致,也与HUNG等[5]的计算结果一致。原子仅在顶部三层产生了明显的弛豫,第四至第六层原子经历了微小的位移,第七至第九层原子的弛豫可以忽略不计,因此可以认为,表面1~3层为表面层,4~6层原子为近体相层,而7~9层原子已经完全具有了体相性质,为体相层。
2.2 Mulliken布居分析
图2所示为计算后表面9层原子的电荷分布。表4所列为各原子层原子的Mulliken电荷布居。由图2可看出,第一层的硫原子带负电荷较大(−0.11 e),由表面至体相,硫原子所带的电荷经历了一个由负到正的过程,体相硫原子带电荷为+0.08 e(S9)。表面铁原子带正电荷(+0.08 e),而表面层以下的近体相及体相铁原子带负电荷,且体相铁原子(Fe12)带最大负电荷(−0.16 e)。由表4列出的轨道布居可知,表面硫原子(S1)主要由p轨道得到电子,s轨道得到少量电子;而S4原子主要由p轨道得到电子,且得到的电子比S1原子的少,因而S1原子拥有最多的负电荷(−0.11 e),而S4原子带少量负电荷(−0.02 e)。表面铁原子主要由p轨道失去电子而带正电荷,d轨道仅失去少量电子。由硫原子和铁原子的电子得失情况可知,在黄铁矿(100)表面上,电荷在铁原子和硫原子之间发生了转移,从铁原子上转移到了硫原子上,这与NESBITT等[26]针对黄铁矿表面提出的观点一致,即在有S—S键断裂的表面上,会发生铁原子失去电子被氧化而硫原子获得电子被还原的反应,而von OERTZEN等[6]采用XPS方法对黄铁矿(100)表面的研究也表明表面电荷从铁原子到硫原子的迁移。此外,原子自旋值表明(100)面上的5配位及体相中的铁原子都是自旋中性的。
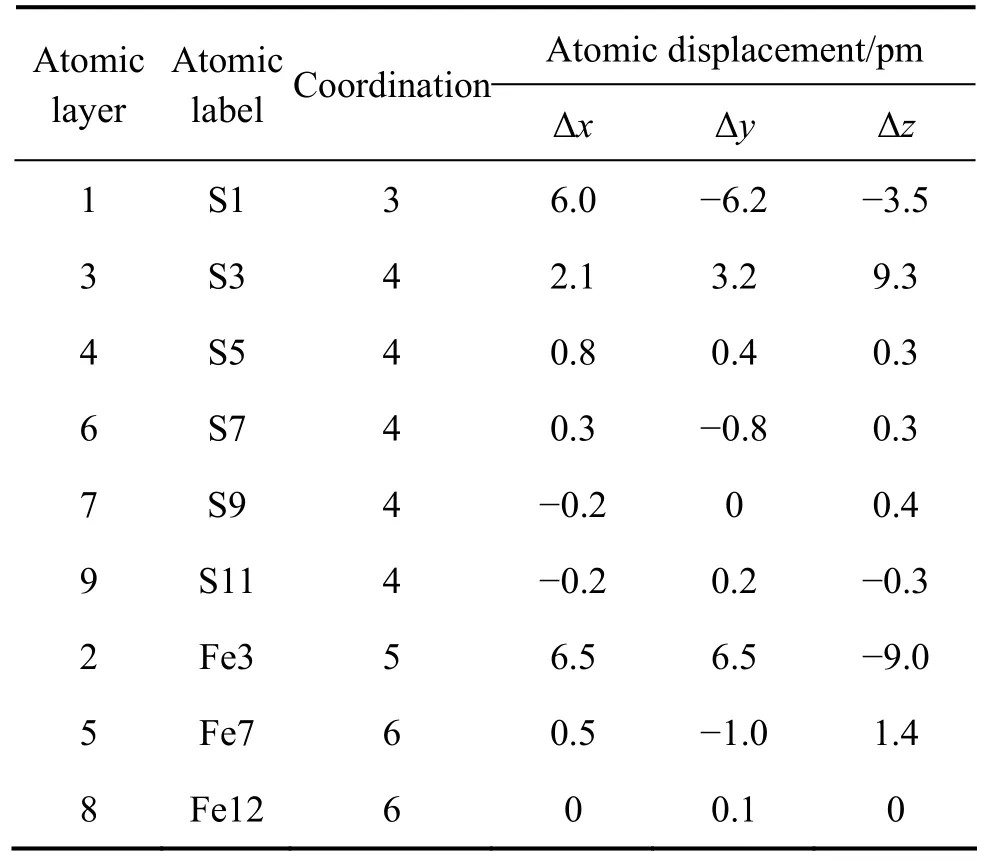
表3 原子配位及位移Table 3 Atomic coordination and displacements
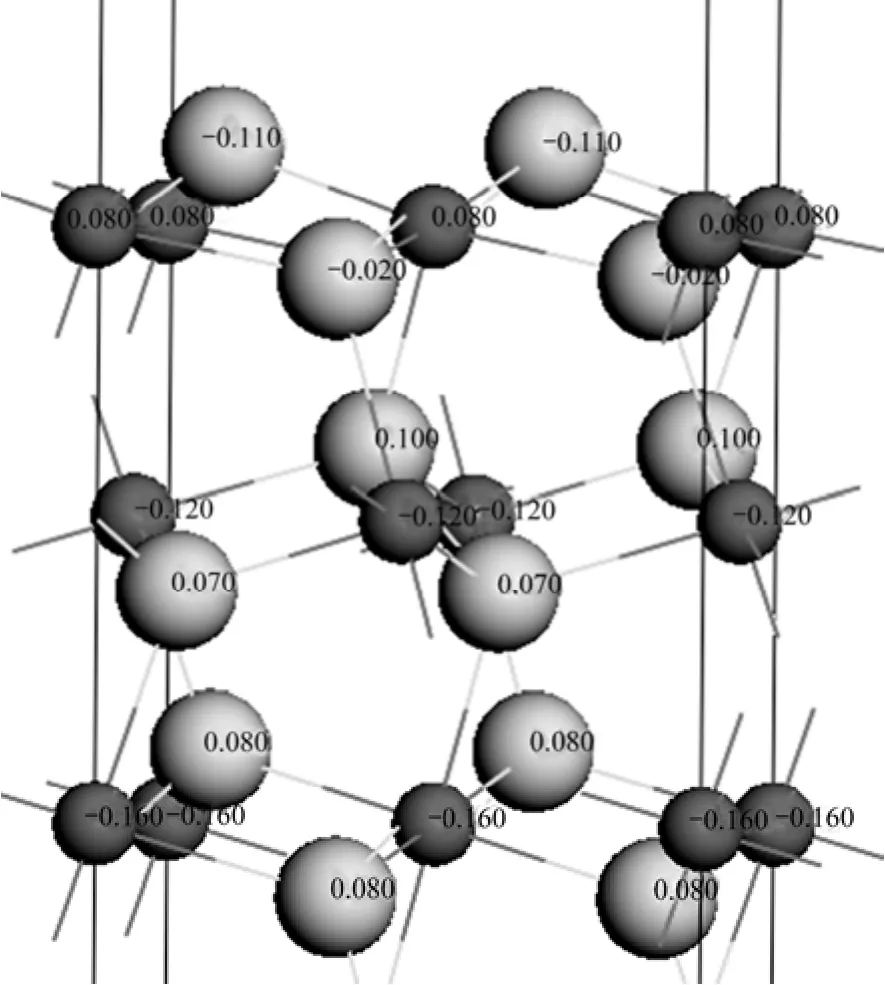
图2 表面9层原子的电荷分布Fig.2 Atomic charge distribution of nine layer of surface
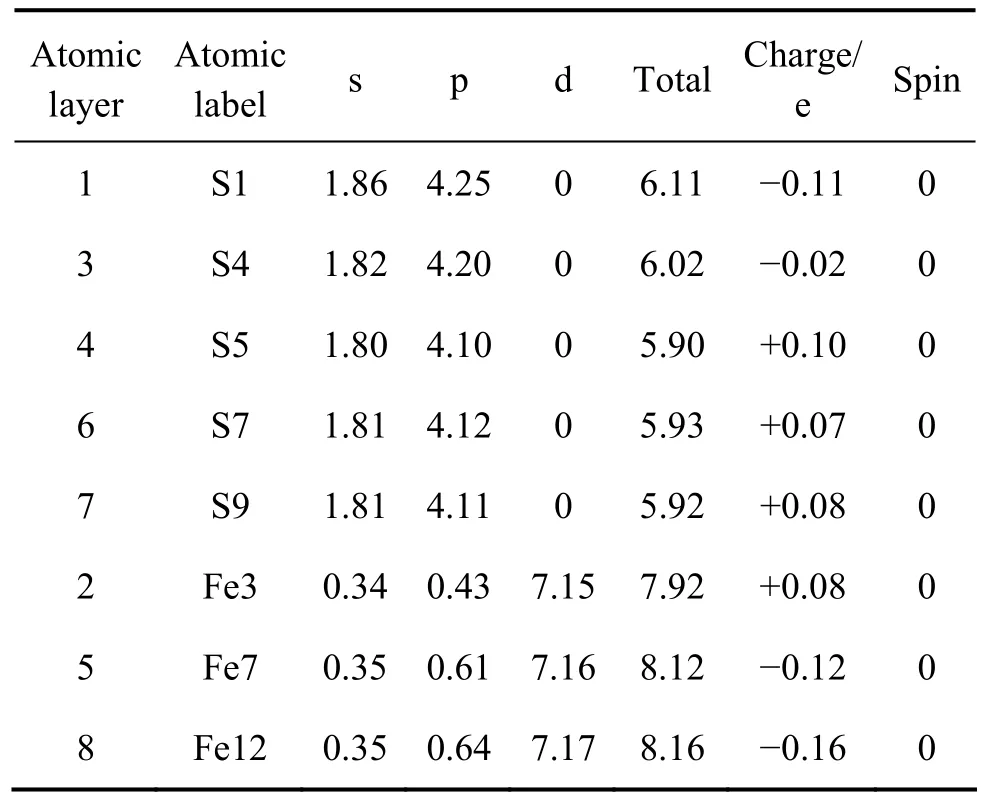
表4 原子的Mulliken电荷布居Table 4 Mulliken charge populations of atoms
从原子的Mulliken电荷布居分析可知,黄铁矿体相中的铁硫原子所带的电荷与实际现象相反,而表面原子却与实际相符,通过对电子差分密度的分析,可以进一步了解存在这种差异的可能原因。由前面对黄铁矿结构的介绍可以知道,在黄铁矿体相中,硫原子与硫原子之间成键,以对硫基团(S)形式存在,这与大多金属硫化物中硫原子只与金属原子成键的现象不同,正是这种差异可能造成了体相中铁和硫原子电荷分布的反常。
图3所示为体相Fe—S、S—S键之间以及表面Fe—S键之间的电荷差分密度。图中蓝色区域表明电子缺失,红色区域表明电子富集,白色表明电子密度几乎没有发生变化的区域。从图3(a)可以看出,电子云在对硫原子之间富集,正是由于两个硫原子之间电子富集,导致强烈的电子排斥作用,并偏向与其配位的铁原子,因此造成硫原子周围的电子缺失,硫原子带正电荷。图3(b)清晰地显示了体相铁原子周围被电子云覆盖。图3(c)显示了表面Fe—S原子之间的电子分布状况。表面Fe3原子靠近真空层一侧由于缺少了一个配位硫原子,电子在这一侧是缺失的,铁原子带正电荷。
另外,从图3(c)可以明显看出,在表面S1上部有明显的电子云富集,且这些电子云由Fe3原子扩散而来,表明电子从表面铁原子向硫原子转移,导致S1原子带负电荷。由此可以看出,表面的计算结果是与实际相符的,同时这也与黄铁矿浮选实践一致,如黄药在黄铁矿表面阳离子的化学吸附作用,以及氢氧根和氰化物与黄铁矿表面铁离子的作用等。另外,对于黄铁矿的电化学无捕收剂浮选,只有表面硫原子带负电荷,才能合理解释黄铁矿表面的硫从负价氧化成零价的元素硫[27]。
Mulliken键的重叠布居可以用来估计键的共价和离子性,较高的布居值表明键呈共价性,而较低的布居值表明键之间的离子相互作用[28−29]。表5所列为相对应的表面和体相原子的键的布居和键长(S1、S2、S3、S4和S5分别与S9、S10、S11、S12和S13的位置相对应)。表面层的键的布居值与体相中的键的布居值存在明显差异,键长也不同。由表5可知,表面Fe3原子与第一层S1和S2原子之间的键的布居值大于体相Fe12原子与S9和S10原子之间键的布居值,表明表面顶部铁−硫原子之间的共价性增强;表面Fe3原子与第三层S3和S4原子之间的布居值小于体相Fe12原子与S11和S12之间的键的布居值,表明它们之间的共价性减弱,而离子性增强;S5—Fe3键之间的布居值与S13—Fe12的接近,这层Fe—S原子之间的共价性强弱变化不大。此外,除S4—Fe3与S12—Fe12之间的键长接近外,表面层Fe—S原子之间的键长都小于体相中相应原子之间的键长,这表明表面上的Fe—S原子之间的相互作用比体相中的更强。

图3 电荷差分密度图Fig.3 Electron density difference map: (a) Two coordinated S atoms; (b) Bulk Fe and its four coordinated S atoms; (c) Surface Fe and its two coordinated S atoms
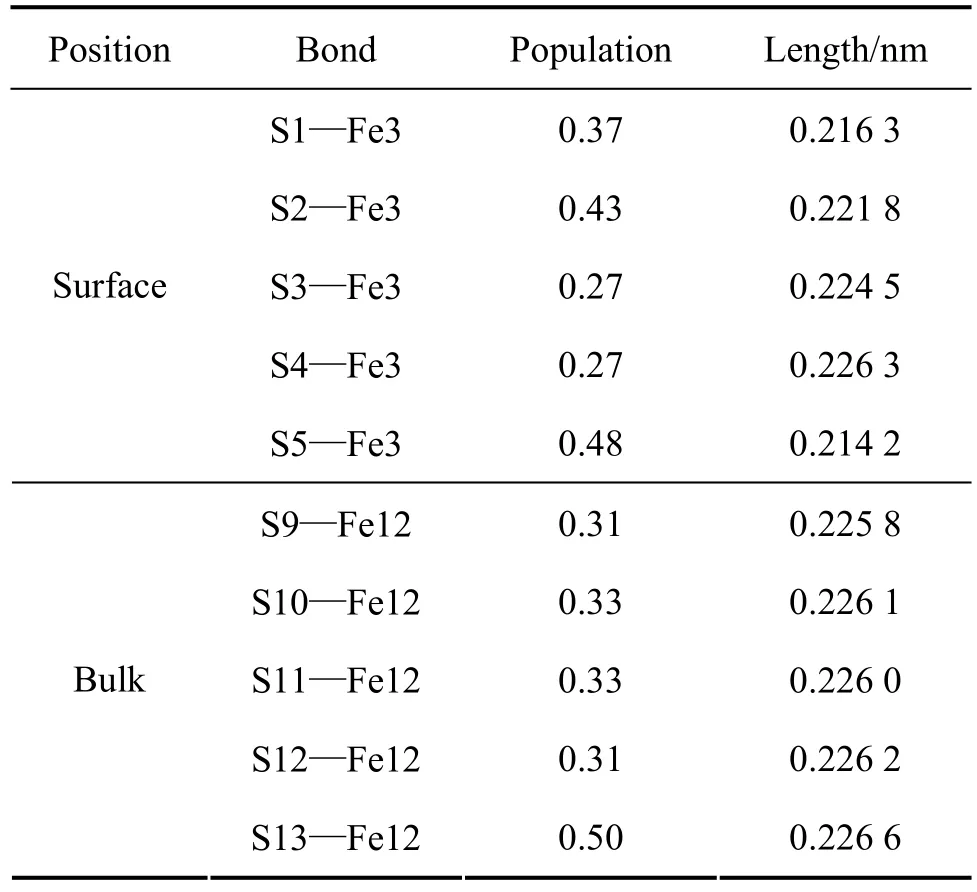
表5 原子的Mulliken键的布居Table 5 Mulliken bond population of atom
2.3 表面电子结构
表面上的原子(Fe3、S1和S4)因所处的环境与体相中的原子不同,所以通过比较它们与近体相及体相原子(F37、S5和S7)的态密度,可以清楚地了解表面电子结构的变化。图4和5分别显示了不同原子层的铁原子和硫原子的态密度。由图4可知(Fe 4s轨道的贡献非常小,未在图中显示),与体相比较,表面铁原子(Fe3)的3d态在−2 eV处的态密度峰强明显增强。而图5的硫原子态密度也显示,与体相硫原子相比,表面S1原子的3p态对−2.5~0 eV之间的能带的贡献明显增强,且−2 eV处的态密度峰强明显增强,Fe—S原子的态密度在这里的重叠增大,表明Fe—S之间的相互作用增强。表面Fe—S原子在0.8 eV处的相互作用也增强了。S5、S7与S9原子的态密度较为相似。
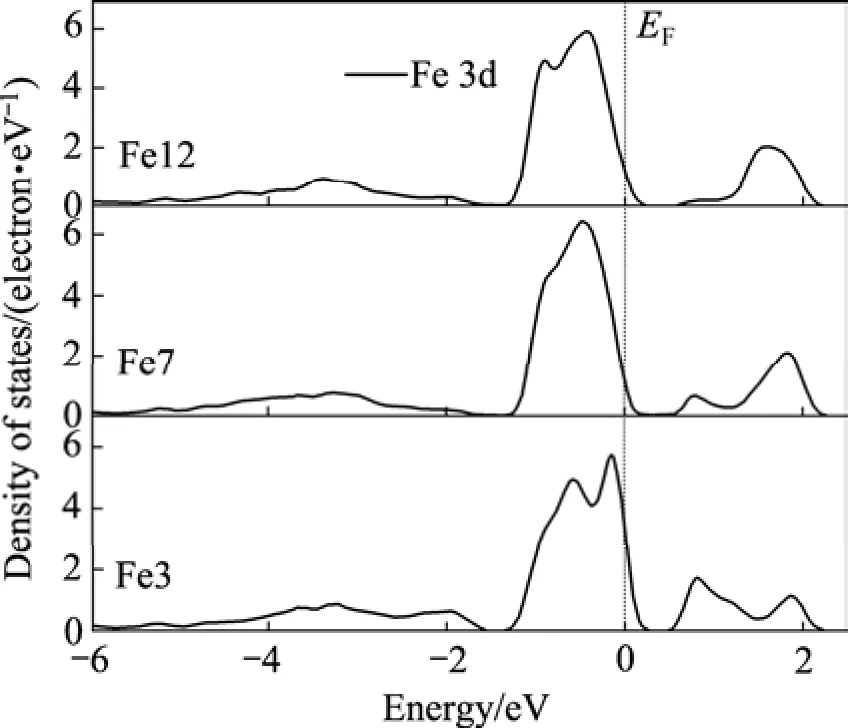
图4 铁原子态密度Fig.4 Density of states of Fe atom
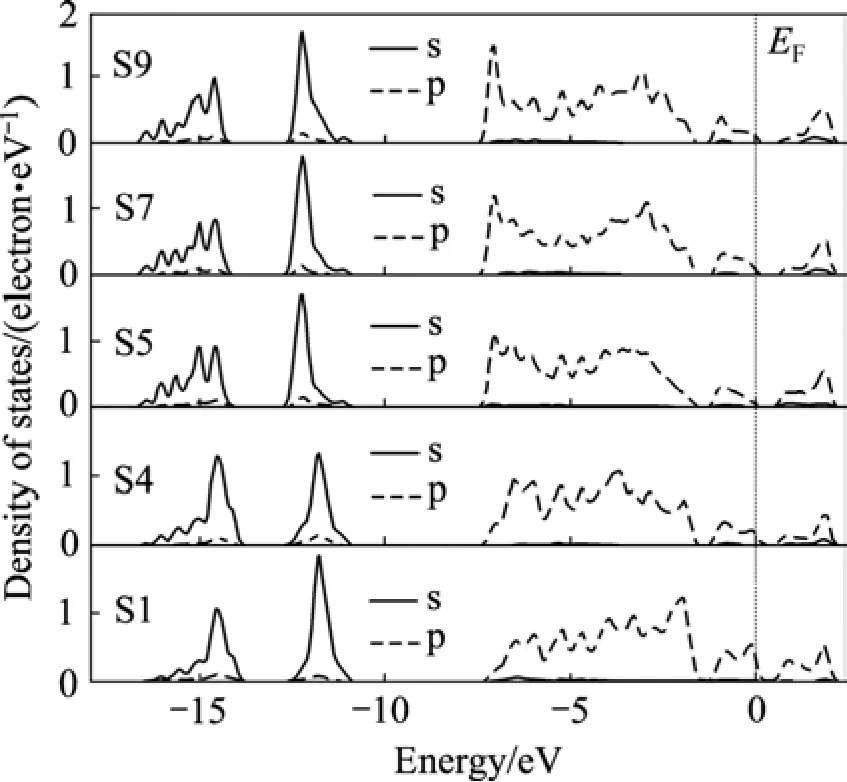
图5 硫原子态密度Fig.5 Density of states of S atoms
在浮选实践中,黄铁矿是最容易受到抑制的硫化矿物,特别是在碱性介质中,黄铁矿可浮性最差,这与黄铁矿表面铁原子的活性有关。由图4可见,黄铁矿表面最外层的铁(Fe3)3d态主要在费米能级附近,这表明黄铁矿表面最外层的铁原子的活性很大,电子给予体容易与表面铁原子发生吸附,因此在碱性介质中黄铁矿容易和氢氧根离子(电子给予体)发生作用,在表面形成氢氧化铁,从而产生抑制作用。
另一方面,从图4和5可以看出,与体相比较,表面铁原子(Fe3)和硫原子(S1)的能隙降低了,且原子态密度明显穿过费米能级(其它原子的态密度稍微穿过费米能级,这是因为smearing宽度的取值的原因),因此黄铁矿(100)表面具有一些似金属特征,这与黄铁矿样品具有光学反射表面的物理外观相一致,也与HUNG等[5]的计算结果一致。
黄铁矿是一种半导体矿物,它的浮选过程涉及电化学反应[30]。传统的电化学理论根据黄铁矿静电位来解释黄药在黄铁矿表面的氧化,即在黄药氧化成双黄药的平衡电位小于黄铁矿的静电位的时候,表面产物为双黄药,反之,则不能氧化成双黄药[31]。静电位理论不能描述出电子在黄铁矿表面的转移情况,同时也不能阐述氧在黄铁矿表面的作用,而电子在黄铁矿表面的转移和氧的作用是黄铁矿浮选电化学过程的重要因素。前面的计算结果表明:当黄铁矿表面解理后,黄铁矿表面层的导电性增强,表面的电化学活性也因此更强。由于黄铁矿表面层具有金属特征而体相层具有半导体特征,一般而言,半导体的费米能级比金属矿物要低,因此,电子有从黄铁矿表面层向体相转移的趋势,从而导致黄铁矿表面具有的吸电子能力。当黄药吸附在黄铁矿表面后,黄药的电子将会向黄铁矿表面转移,并向体相传递,氧化成双黄药,这与黄铁矿有电催化能力的实际相符合。当黄铁矿表面只存在黄药的时候,在电子从表面向体相转移过程中会形成肖特基势垒,阻止电子的传递,从而阻碍黄药的进一步氧化,因此,在无氧条件下黄铁矿表面难以形成较多的双黄药,不利于黄铁矿的浮选。当黄铁矿表面吸附氧后,由于氧对电子的亲和能力远大于黄铁矿的,因此黄铁矿表面的电子会向氧转移,而不是向体相转移,在这种环境下,就形成了黄铁矿与黄药反应的电化学共轭过程[32],即黄药在黄铁矿表面失去电子发生阳极反应形成双黄药,而氧分子则从黄铁矿表面得到电子发生阴极反应,从而有利于黄铁矿的浮选。
3 结论
1) 黄铁矿(100)面经历较小的弛豫,且弛豫主要发生在表面配位不足的铁原子周围,表面未发生明显的重构现象。
2) 铁原子的电荷经历了一个由表面到体相从正到负的过程,而硫原子则经历了从负到正的过程,电子在表面铁原子和硫原子之间转移,由表面铁原子转移到表面硫原子上。
3) 表面铁−硫原子之间的相互作用强于体相中的铁−硫原子。
4) 表面五配位的铁原子是自旋中性的;表面铁原子的活性很大,因此在碱性介质中黄铁矿容易和氢氧根离子(电子给予体)发生作用,在表面形成氢氧化铁,从而产生抑制作用。
5) 黄铁矿(100)表面层的能隙降低了,导电性相对于体相增强,表面带有一些似金属特征,表面的电化学活性也增强了,电子有从黄铁矿表面层向体相转移的趋势,并在这些计算结果的基础上建立了黄药和氧气与黄铁矿表面作用的电化学模型。
REFERENCES
[1] LAAJALEHTO K, KARTIO I, SUONINEN E. XPS and SR-XPS techniques applied to sulphide mineral surfaces[J].International Journal of Mineral Processing, 1997, 51(1):163−170.
[2] ROSSO K M, BECKER U, FOCHELLA M F. Atomically resolved electronic structure of pyrite {100} surfaces: An experimental and theoretical investigation with implications for reactivity[J]. American Mineralogist, 1999, 84(10): 1353−1548.
[3] CHATURVEDI S, KATZ R, GUEVREMONT J, SCHOONEN M A A, STRONGIN D R. XPS and LEED study of a single-crystal surface of pyrite[J]. American Mineralogist, 1996,81(1/2): 261−264.
[4] LEIRO J A, MATTILA S S, LAAJALEHTO K. XPS study of the sulphur 2p spectra of pyrite[J]. Surface Science, 2003, 547(1):157−161.
[5] HUNG A, MUSCAT J, YAROVSDY I, RUSSO S P.Density-functional theory studies of pyrite FeS2(100) and (110)surfaces[J]. Surface Science, 2002, 513(3): 511−524.
[6] von OERTZEN G U, SKINNER W M, NESBITT H W. Ab initio and XPS studies of pyrite (100) surface states[J]. Radiation Physics and Chemistry, 2006, 75(11): 1855−1860.
[7] CAI J, PHILPOTT M R. Electronic structure of bulk and (001)surface layers of pyrite FeS2[J]. Computational Materials Science, 2004, 30(3/4): 358−363.
[8] EDELBRO R, SANDSTR Ö M Å, PAUL J. Full potential calculations on the electron bandstructures of sphalerite, pyrite and chalcopyrite[J]. Applied Surface Science, 2003, 206(1/4):300−313.
[9] von OERTZEN G U, JONES R T, GERSON A R. Electronic and optical properties of Fe, Zn and Pb sulfides[J]. Physics and Chemistry of Minerals, 2005, 32(4): 255−268.
[10] WONES M, KARNATAK R C, ESTEVA J M, LEFEBVRE I,ALLA G, OLIVIER-FOURCADE J, JUMAS J C. Electronic structures of FeS and FeS2: X-ray absorption spectroscopy and band structure calculations[J]. Journal of Physics and Chemistry of Solids, 1997, 58(2): 345−352.
[11] von OERTZEN G U, SKINNER W M, NESBITT H W. Ab initio and x-ray photoemission spectroscopy study of the bulk and surface electronic structure of pyrite (100) with implications for reactivity[J]. Physical Review B, 2005, 72(23): 235427-1−235427-10.
[12] OPAHLE I, KOEPERNIK K, ESCHRIG H. Full potential band structure calculation of iron pyrite[J]. Computational Materials Science, 2000, 17(2/4): 206−210.
[13] 李玉琼, 陈建华, 陈 晔. 空位缺陷黄铁矿的电子结构及其浮选行为[J]. 物理化学学报, 2010, 26(5): 1435−1441.LI Yu-qiong, CHEN Jian-hua, CHEN Ye. Electronic structures and flotation behavior of pyrite containing vacancy defects[J].Acta Physico-Chimica Sinica, 2010, 26(5): 1435−1441.
[14] HUNG A, MUSCAT J, YAROVSKY I, RUSSO S P.Density-functional theory studies of pyrite FeS2(111) and (210)surfaces[J]. Surface Science, 2002, 520(1): 111−119.
[15] von OERTZEN G U, SKINNER W M, NESBITT H W, PRATT A R, BUCKLEY A N. Cu adsorption on pyrite (100): Ab initio and spectroscopic studies[J]. Surface Science, 2007, 601(24):5794−5799.
[16] 王淀佐, 龙翔云, 孙水欲. 硫化矿的氧化与浮选机理的量子化学研究[J]. 中国有色金属学报, 1991, 1(1): 15−23.WANG Dian-zuo, LONG Xiang-yun, SUN Shui-yu. Quantum chemical study on the mechanism of oxidation and flotation of sulfide minerals[J]. The Chinese Journal of Nonferrous Metals,1991, 1(1): 15−23.
[17] CLARK S J, SEGALL M D, PICKARD C J, HASNIP P J,PROVERT M I J, REFSON K, PAYNE M C. First principles methods using CASTEP[J]. Zeitschrift fuer Kristallograhie, 2005,220(5/6): 567−570.
[18] SEGALL M D, LINDAN P J D, PRONBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: ideas, illustrations and the CASTEP code[J]. Journal of Physics: Condensed Matter, 2002, 14(11): 2717−2744.
[19] 谢希德, 陆 栋. 固体能带理论[M]. 上海: 复旦大学出版社,1998: 1−26.XIE Xi-de, LU Dong. Energy band theory of solids[M].Shanghai: Fudan University Press, 1998: 1−26.
[20] MARZARI N, VANDERBILT D, PAYNE M C. Ensemble density-functional theory for ab-initio molecular dynamics of metals and finite-temperature insulators[J]. Physical Review Letters, 1997, 79(7): 1337−1340.
[21] JONES R O, GUNNARSSON O. The density functional formalism, its applications and prospects[J]. Reviews of Modern Physics, 1989, 61(3): 689−746.
[22] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects[J]. Physical Review, 1965,140(4A): A1133−A1138.
[23] MONKHORST J, PACK J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 13(12): 5188−5192.
[24] PCAK J D, MONKHORST H J. Special point for Brillouin-zone integrations—A reply[J]. Physical Review B, 1977, 16(4):1748−1749.
[25] VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Physical Review B, 1990,41(11): 7892−7895.
[26] NESBITT H W, BANCROFT G M, PRATT A R, SCAINI M J.Sulfur and iron surface states on fractured pyrite surfaces[J].American Mineralogist, 1998, 83(9/10): 1067−1076.
[27] 杨梅金, 陈建华, 马少键, 胡治流. 黄铁矿电化学浮选研究[J].广西大学学报: 自然科学版, 2002, 27(4): 281−283.YANG Mei-jin, CHEN Jian-hua, MA Shao-jian, HU Zhi-liu.Research of electrochemical flotation of pyrite[J]. Journal of Guangxi University: Natural Science Edition, 2002, 27(4):281−283.
[28] SEGALL M D, SHAH R, PICKARD C J, PAYNE M C.Population analysis in plane wave electronic structure calculations[J]. Molecular Physics, 1996, 89(2): 571−577.
[29] SEGALL M D, SHAH R, PICKARD C J, PAYNE M C.Population analysis of plane-wave electronic structure calculations of bulk materials[J]. Physical Review B, 1996,54(23): 16317−16320.
[30] 陈建华, 冯其明, 卢毅屏. 电化学调控浮选能带模型及应用(Ⅱ)—黄药与硫化矿物作用的能带模型[J]. 中国有色金属学报, 2000, 10(3): 426−429.CHEN Jian-hua, FENG Qi-ming, LU Yi-ping. Energy band model of electrochemical flotation and its application (Ⅱ)—Energy band model of xanthate interacting with sulphide minerals[J]. The Chinese Journall of Nonferrous Metals, 2000,10(3): 426−429.
[31] ALLISON S A, GOOLD L A, NICOL M J, GRANVILLE A. A determination of the products of reaction between various sulfide minerals and aqueous xanthate solution, and a correlation of the products with electrode rest potentials[J]. Metallurgical and Materials Transactions B, 1972, 3(10): 2613−2618.
[32] 冯其明, 陈 荩. 硫化矿物浮选电化学[M]. 长沙: 中南工业大学出版社, 1992: 70−79.FENG Qi-ming, CHEN Jin. Electrochemistry of sulfide mineral flotation[M]. Changsha: Central South University of Technology Press, 1992: 70−79.
Density functional theory calculation of surface properties of pyrite (100) with implications for flotation
LI Yu-qiong1, CHEN Jian-hua2,3, CHEN Ye2, GUO Jin3
(1. School of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China;
2. School of Resources and Metallurgy, Guangxi University, Nanning 530004, China;3. School of Physics Science and Engineering, Guangxi University, Nanning 530004, China)
The structural relaxation, atomic Mulliken populations and electronic structures of ideal pyrite (100) surface were calculated using density functional theory (DFT) method of plane-wave pseudopotential method. The reason for the charge unconventionality in bulk pyrite was explained. Moreover, the effects of structures and properties of surface on pyrite flotation behavior were analyzed from a flotation point of view. The calculated results show that the relaxation of pyrite (100) surface is relatively small, and the Fe-S interaction increases at the surface compared to that in the bulk. The calculated electronic structure results suggest that the surface 5-coordinated Fe atom has high activity. The energy gap of surface Fe and S atoms decreases, suggesting higher conductivity, as well as higher electrochemical activity, of the surface layer than the bulk.
pyrite surface; density functional theory calculation; Mulliken populations; electronic structures; flotation
TD923
A
1004-0609(2011)04-0919-08
国家自然科学基金资助项目(50864001)
2010-04-15;
2010-12-28
陈建华,教授,博士;电话:0773-3232200;E-mail: jhchen1971@sina.com
(编辑 李艳红)
