海水电池用镁阳极的研究与应用
2011-11-03王日初彭超群
冯 艳,王日初,彭超群
(中南大学 材料科学与工程学院,长沙 410083)
海水电池用镁阳极的研究与应用
冯 艳,王日初,彭超群
(中南大学 材料科学与工程学院,长沙 410083)
镁阳极材料具有电负性好,比能量高以及密度小等优异性能,在海水电池领域具有广阔的应用前景。综述了近年来国内外镁阳极海水电池的研究及应用,讨论镁阳极材料的活化机制和腐蚀行为,探讨合金元素和第二相对镁阳极电化学活性和耐腐蚀性能的影响,指出今后海水电池用镁阳极材料的发展应在充分研究合金元素活化机理和第二相影响的基础上,研制出自腐蚀速率更小、阳极利用率更高以及比能量更大的新型镁合金阳极材料。关键词:海水电池;镁阳极;活化;腐蚀;合金元素;第二相
海水电池是在第二次世界战争期间由美国贝尔实验室设计、通用电气公司研制的,它依靠阳极金属材料在海水中的腐蚀溶解提供阳极放电电流,而阴极则主要依靠海水中的溶解氧在惰性的气体电极上进行还原反应提供阴极电流。海水电池最突出的特点是不需要携带电解质,可以在需要的时候利用天然海水形成电解液。出于不同使用目的,海水电池具有多种不同类型,如大功率水下武器装备的动力电池,长周期、小功率的水中探测仪器类电池以及水下航行体的动力电池——半燃料海水电池等[1]。其中大功率动力电池的应用前景最广,技术难度最大,研制也最有战略意义,目前制约其性能的重要一方面就在阳极材料的开发上[2]。自从20世纪40年代以来,美国和一些发达国家的政府和商业机构就已经开始研究和研制在海水中的大功率动力电池[3−5]。
成功应用在大功率海水电池中的阳极材料集中在镁合金和铝合金上[3−7],这主要是由于镁、铝具有优于其他金属的阳极性能,如电极电位负、密度小、比容量高等。常用的阳极金属锂电位最负,为−3.05 V,但化学性质过于活泼,无法用于水溶液类电解液电池,至今锂由于安全性差而无法应用于大功率放电。事实上,镁、铝自身也有作为阳极材料不可回避的缺陷。铝比能量高于镁的,但其阳极电位在同等条件下低于镁的,应用于海水电池时其反应产物为絮状沉淀物Al(OH)3,易造成腐蚀产物堆积而影响电池性能,同时由于铝是两性金属,容易与介质发生严重析氢反应[7]。镁具有较高的电化学活性,阳极电位为−2.363 V,电化学当量为2 200 A·h/kg,仅低于锂和铝的[8]。纯镁表面的微观腐蚀电池驱动力大,易发生微观腐蚀原电池反应,产生大量氢气,导致阳极的法拉第效率降低。20世纪60~80年代,国外已对镁阳极进行了广泛的研究与试验,普遍采用合金化的方法对镁阳极进行性能优化。研究结果表明,通过合金化的镁合金能够在较大电流密度下减小自放电,得到60%的效率,比铝合金更适合作为冷的海水溶氧电池的阳极[1]。目前已开发的应用于大功率海水电池的镁阳极材料有英国镁电子公司生产的AP65和MT75以及俄罗斯和国内中南大学研制的Mg-Hg阳极材料[8],具体成分和制备工艺未见报道。
镁合金阳极虽然在大功率海水电池中得到应用,但现有的镁合金阳极材料仍存在自腐蚀速度大、阳极利用率低等不足之处,寻找阳极利用率高的镁合金阳极材料是国际上镁电池研究的热点和难点问题。对镁阳极的合金化元素的设计需要人们对合金元素作用的认识进一步提高,如Zn、Bi、Pb和In等,其活化机理仍没有统一的解释[9]。本文作者从活化机制、腐蚀行为、合金元素以及第二相对海水电池用镁阳极性能的影响等方面,探讨海水电池用镁阳极的发展方向。
1 常用镁阳极海水电池
1.1 镁/氯化银海水电池
镁/氯化银海水电池采用金属 Mg做阳极,AgCl做阴极,反应原理如下[1]:
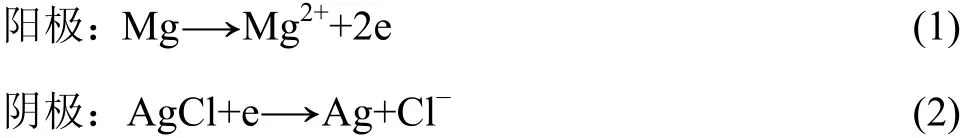
镁阳极在海水中能长期保持其活性,因为氯化物海水是镁阳极很好的活化溶液,同时由于镁的极化较大,因此电极反应的热效应较大,这一热量保证了该电池具有良好的低温性能,无需辅助加热装置就可适应−60 ℃低温;选用溶解度低的 AgCl作为阳极,AgCl/Ag电位非常稳定,能作为中性溶液中的参比电极使用,其放电后转化为导电性良好的 Ag,减少Mg/AgCl海水电池内阻,使其适宜于大电流密度下工作,比能量可达88 W·h/kg[10−11]。由于靠海水激活,因此Mg/AgCl海水电池平时处于干态保存,搁置时间可长达5 a,但这一体系需要消耗贵金属Ag,造价高,且总功率有待提高。
Mg/AgCl海水电池作为一次激活贮备电池,采用双极性堆式结构[12]。电池的阳极为含少量 Al、Zn和Pb等元素的合金,与纯Mg电池相比,其比能量提高20%。这种电池20世纪60年代开始在美国的MK44鱼雷、MK45鱼雷上使用,迄今已有40多年的历史。目前正在服役的意大利A244 /S鱼雷、英国的“鯆鱼”鱼雷、法国的 R3鱼雷都以这种电池为动力[13],但电池结构不同,性能各异,其性能如表1所列[14−15]。
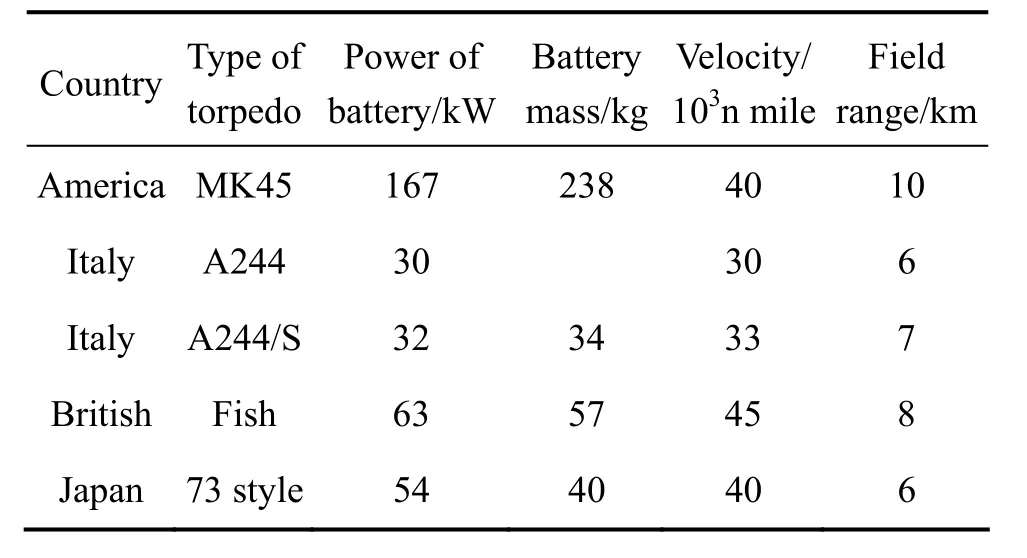
表1 各国鱼雷动力电池性能[14−15]Table 1 Performances of battery for torpedo in many countries[14−15]
1.2 镁/氯化亚铜海水电池
这是由前苏联开发的海水电池,目前俄罗斯仍然大量使用。该电池的阴极材料采用相对经济的铜合金材料代替贵金属银。为防止其氧化,加入一定数量的SnCl2,同时采用氩气保护措施,保证电极的活性。采用镁汞合金作阳极,汞齐化主要是提高镁的稳定性和表面的析氢过电位,抑制镁阳极的自腐蚀。镁/氯化亚铜海水电池电极反应原理如下:
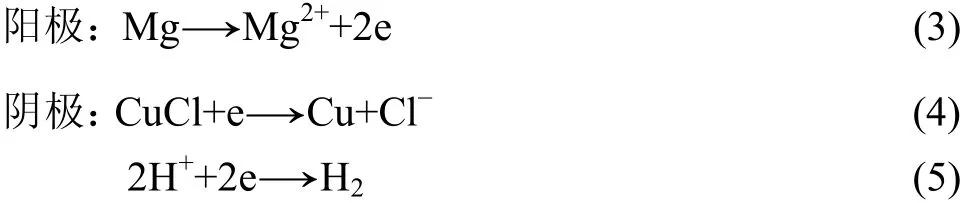
该电池造价只有Al/AgO海水电池的1/3,但电池体积较大,目前只有俄罗斯实际应用到 y ∋T T 、TT−80型鱼雷上。王日初等[16]也对这种电池的阳极材料做了大量研究,开发了Mg-Hg-Ga阳极材料。研究表明,这种阳极材料在 200 mA/cm2的电流密度下,平均电压达到−1.8 V,阳极极化小,具有很好的电化学活性。
2 海水电池用镁阳极的活化机理
迄今为止,对镁阳极活化机理的研究还大多数集中在二元和三元合金上,而且没有得出统一观点,然而现在镁合金阳极已经发展到五元甚至七元[17],其反应机理也变得更加复杂。因此,在研究镁合金阳极活化机理时,镁合金的微观组织、镁合金中各元素的相互作用以及合金元素与海洋环境中 Cl−电解液之间的相互作用是都镁合金阳极活化的关键。
冯艳等[16]提出含Hg、Ga元素的镁阳极活化机理:溶解—再沉积。他们认为:在溶解初期,阴极性第二相化合物通过腐蚀原电池反应引发镁基体点蚀的出现,促进了Mg基体和合金元素Hg、Ga的溶解;在放电过程中,溶液中的Hg+和Ga3+被Mg还原成Hg、Ga而沉积在阳极表面,反应原理如下:

Hg和Ga沉积层一方面形成镁汞齐,镁汞齐与水发生剧烈反应而继续产生Hg和Ga沉积,此循环促进活化,反应式如下:

Hg,Ga沉积层另一方面影响镁阳极表面结构(见图1),隔离腐蚀产物,破坏氧化物膜,从而使合金电位负移,点蚀更易引发和扩散,进一步起到活化作用。

图1 Mg-Hg-Ga阳极放电过程的表面结构示意图Fig.1 Schematic diagram of surface structure of Mg-Hg-Ga anode during electric
3 海水电池用镁阳极的腐蚀
3.1 腐蚀类型
从腐蚀形貌来看,镁阳极的腐蚀并不是均匀腐蚀,其不均匀程度与合金的种类及腐蚀环境有关[18],海洋环境中的 Cl−使镁阳极局部区域置于酸性环境中,加速腐蚀[19−20]。在很多情况下,镁阳极的腐蚀一般都是从某一局部腐蚀开始的,常常表现为点蚀,其腐蚀小孔可深可浅,然后发展开来。一般来说,点蚀、电偶腐蚀和晶间腐蚀是镁阳极材料中最常见和最重要的腐蚀类型[21−22]。
3.1.1 点蚀
在镁合金阳极表面有一层氧化膜,氧化膜的致密度为氧化物分子体积与形成该氧化物的金属原子体积之比[23],镁的致密度小于 1,说明其氧化物膜疏松、多孔,存在缺陷,容易造成点蚀。另外,第二相化合物和基体之间的微电池反应也是产生点蚀的原因之一。在微电池反应中,第二相为阴极,基体为阳极,在第二相周围的基体发生溶解,最后导致第二相脱落,从而产生点蚀坑[24]。图2所示为Mg-Hg-Ga阳极材料蚀坑的截面形貌[8]。

图2 Mg-Hg-Ga合金在NaCl溶液中蚀坑的截面形貌[8]Fig.2 Morphology of cross-section of pit in Mg-Hg-Ga alloys in NaCl solution[8]
图3 所示为镁合金表面点蚀发展示意图。以此说明点蚀发展的过程[8]。当点蚀发生后,点蚀坑底部金属镁便发生溶解,即 Mg→Mg2++2e,孔内金属离子不断增加。在含有氯离子的水溶液中,在蚀孔电池产生的电场作用下,蚀孔外阴离子(Cl-)不断向孔内迁移、富集,孔内氯离子浓度升高,同时由于孔内金属离子浓度升高并发生水解:Mg2++2H2O→Mg(OH)2+2H+,使孔内溶液氢离子浓度升高,pH值降低,溶液酸化,相当于使蚀孔内金属处于HCl介质中,处于活化溶解状态。水解产生的氢离子和孔内的氯离子又促使蚀孔侧壁的镁继续溶解,发生自催化反应:Mg+2H+→Mg2++H2。由于孔内浓盐溶液中氧的溶解度很低,又加上扩散困难,使得闭塞电池局部供氧受到限制,孔内的镁难以再形成氧化膜,而一直处于活化状态。蚀孔口形成的Mg(OH)2腐蚀产物沉积层,阻碍了扩散和对流,使孔内溶液得不到稀释,从而造成上述电池效应。
3.1.2 电偶腐蚀
镁阳极中不仅存在不同的相、杂质与缺陷,即使是同一基体相,其不同部位合金元素的含量也不同,故镁合金表面是不可能电化学均匀的,因此镁阳极易由于上述微观结构特征引起局部电偶腐蚀。具体来说,镁阳极中引起腐蚀微电偶的原因有以下几种。
1) 成分不均匀
在镁阳极基体相中,成分的分布并不均匀,如初生的基体相与共晶的基体相或包晶的基体相在合金元素的含量上就有很大不同。这些成分上的不同,导致镁阳极表面电化学行为不同,因此,镁阳极在基体相中会形成电偶腐蚀。
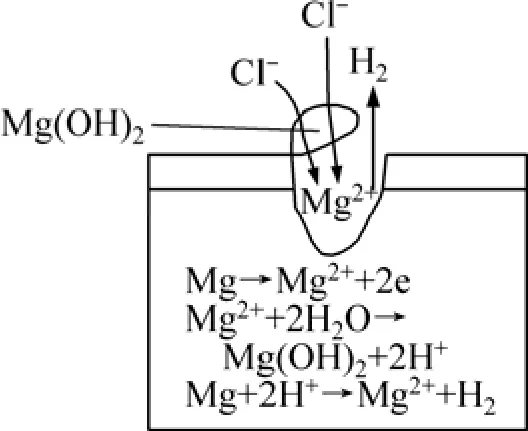
图3 镁合金点蚀发展示意图[8]Fig.3 Schematic diagram of pitting development of Mg alloys[8]

图4 Mg-Hg-Ga合金时效不同时间后第二相的形貌Fig.4 Morphologies of second-phases in Mg-Hg-Ga alloy after aging for different times: (a) 1 h; (b) 200 h
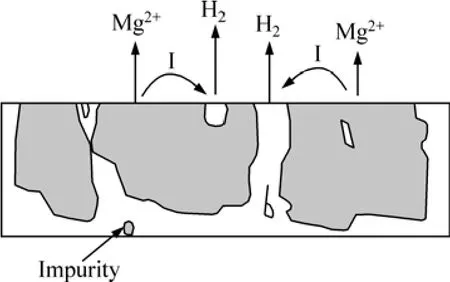
图5 杂质元素引起镁合金的电偶腐蚀示意图[28]Fig.5 Schematic diagram of electric corrosion of Mg alloy due to impurity[28]
2) 第二相
为了获得更高电化学活性和电流利用率的镁阳极,一般在镁中添加各种合金元素形成第二相[25],如Mg-Hg-Ga合金中的Mg3Hg和Mg5Ga2,Mg-Pb合金中的Mg2Pb,Mg-Sn合金中的Mg2Sn,Mg-Sr合金中的Mg9Sr,Mg-Zn合金中的Mg2Zn等。镁阳极中的这些第二相,其自腐蚀电位一般都比基体相高。有人定义了一个驱动力的概念来描述合金元素相对镁腐蚀速率的影响[26],即在镁合金中,某一相的腐蚀驱动力应该等于该相的电位与镁的电位差,再减去该相的析氢过电位。虽然这是一个极其粗略的评估第二相对基体相电偶效应大小的方法,但它实际上已经考虑了第二相对基体相电偶效应的主要因素。
事实上,第二相对镁基体的腐蚀极大取决于镁阳极合金中第二相的数量和分布。以Mg-Hg-Ga为例,图4所示为同一成分的Mg-Hg-Ga合金时效不同时间后的微观形貌[27],EDS和XRD分析显示,图4(a)中Mg3Hg和α-Mg形成共晶组织,呈网状分布在晶界,而图4(b)中的第二相Mg3Hg则成块状均匀弥散地分布在整个晶体中。由这两个合金的动电位极化曲线扫描结果可知,当第二相Mg3Hg与基相α-Mg形成共晶在晶界呈网状分布时其腐蚀电流密度为70.92 mA/cm2,而第二相 Mg3Hg在晶内呈块状均匀弥散分布时其腐蚀电流密度为 2.34 mA/cm2,比网状共晶分布时小很多。这说明阴极性第二相Mg3Hg均匀弥散分布时,可对基体 α-Mg的腐蚀起到阻碍作用,提高 Mg-Hg-Ga阳极的耐腐蚀性能[27]。
3) 杂质元素
一般来说,杂质元素及其化合物具有比镁基体相正得多的电极电位,而且,这些杂质的阴极析氢过电位都很低,与镁合金构成微电池时,它们都是十分有效的阴极相,能极大地促进镁合金的电偶腐蚀。这是杂质对镁合金腐蚀的直接加速作用,其腐蚀作用示意图如图5所示[28]。
固溶于镁合金中的杂质虽然对镁合金的腐蚀影响不大,但可能随镁合金的腐蚀而溶解到溶液中,而后再被镁还原成单质沉积回镁的表面形成腐蚀电偶,促进其沉积处镁合金的进一步腐蚀。这种不利作用是杂质的二次影响。
4) 由局部腐蚀引发的电偶腐蚀
不仅上述的这些镁阳极本身的微观组织形成的微电偶导致电偶腐蚀的发生,镁阳极中局部腐蚀的发生本身也是一种腐蚀微电偶。例如,腐蚀过程中先发生的点蚀导致表面膜破裂,这些破裂处的表面与表面膜未破裂处的金属存在电化学差异,从而导致电偶腐蚀发生,使得镁阳极表面的腐蚀进一步扩展。
3.1.3 晶间腐蚀
晶间腐蚀是指腐蚀沿着金属或合金的晶粒边界或它的邻近区域发展;而晶粒本身的腐蚀是很轻微的一种腐蚀类型,它是由晶粒与晶界之间的电位差引起的局部腐蚀。镁合金阳极由于其较高的电化学活性,置于海洋性大气环境等腐蚀性强的介质中时,有时会发生晶间腐蚀。这种腐蚀使晶粒间结合力大大降低,降低了镁阳极材料的电流效率。
含有不同合金元素的镁阳极,其晶间腐蚀的倾向不同。例如含铝的镁阳极,由于铝在晶粒内部含量低于晶粒周边的,一般是晶粒内部先被腐蚀,其发生晶间腐蚀的倾向很小,但若Mg、Al形成大量的MgAl、Mg2Al3、Mg4Al3等金属间化合物[25],这些金属间化合物在晶间的存在,会引起晶间腐蚀,增大镁的自腐蚀速度,加速固溶体的破坏;含锆的镁阳极晶界上的第二相是相对稳定的,合金元素锆主要分布于晶内而发生晶间腐蚀;Mg-1%Ga(质量分数)合金由于晶界生成Mg5Ga2相,腐蚀沿着晶粒边界或它的邻近区域发生,而晶粒本身腐蚀很轻微,图6所示为Mg-1%Ga合金的晶界腐蚀形貌。

图6 Mg-1%Ga合金的晶界腐蚀形貌Fig.6 Morphology of grain-boundary corrosion in Mg-1%Ga alloy
3.2 腐蚀机理
3.2.1 总腐蚀反应
镁合金的腐蚀性由合金中单个组元相的腐蚀反应控制,如果合金中含有与环境反应激烈的组元,则耐腐蚀性能很差。纯镁的腐蚀反应很重要,提供理解各种镁合金腐蚀的基础。
镁在水溶液环境中溶解是发生与水的电化学反应,这个反应产生氢氧化镁和氢气,因此镁的腐蚀对氧气的浓度不敏感[29−31],然而氧气的存在在空气腐蚀中是很重要的因素[32]。镁在水溶液环境中的腐蚀通常包括阴阳两极之间的微电偶腐蚀[33]。

阳极反应式(10)还包括产生存在时间不长的单价镁离子(Mg+)的中间步骤[30,34]。氢离子的还原过程和阴极相的析氢过电位在镁的腐蚀中起重要作用,低析氢过电位的阴极相使析氢便利,产生较大的腐蚀速率[32]。
目前,关于镁合金腐蚀反应的系统研究还没有报道。有研究表明,镁合金的腐蚀反应类似于纯镁的反应。例如,SONG等[35−36]研究了 Mg-Al-Zn合金的阳极溶解,结果表明Mg是溶液中溶解的主要组元,Al发生少量溶解,没有发现 Zn在溶液中溶解。这意味着对于Mg-Al-Zn合金来说,反应式(9)~(12)的4个反应可以反映其主要的腐蚀过程,当然,也不能排除合金元素Al、Zn对镁腐蚀反应速率的作用。
3.2.2 负差数效应
一般而言,铁、铜合金在酸性溶液中随着极化电位的升高,其阳极溶解率增加,阴极析氢速率降低,但镁合金的析氢腐蚀反而增加。这种行为看似与电化学原理相冲突,被定义为负差数效应,见图 7[37]。从曲线A-B-F-C可知,极化电位从负变到正值时,镁合金的析氢速率开始降低,然后升高。
SONG等[28]根据对实验数据的仔细分析,提出了一种新的“阳极析氢”的腐蚀机制来解释负差数效应,见图8。
镁合金的表面都存在一层白色的 Mg(OH)2保护膜层,但是这层膜并不完整致密,在镁合金服役期间,这层膜的不完整性随着极化电位的升高而加大,正是这层表面膜的破裂对镁合金的腐蚀起着决定性的作用。
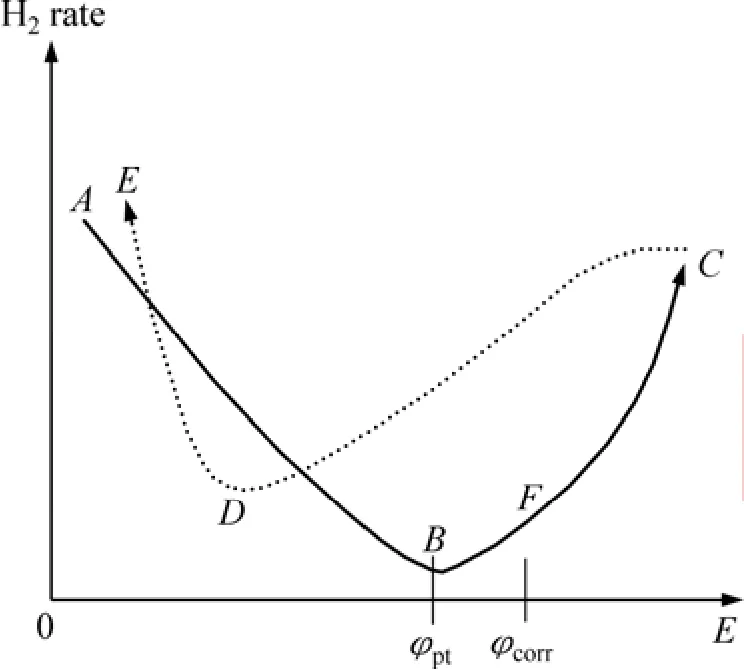
图7 镁合金析氢速率与极化电位关系示意图[37]Fig.7 Schematic diagram of dependence of hydrogen evolution rate from polarization magnesium alloy[37]
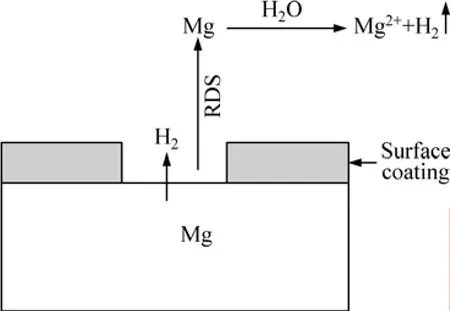
图8 镁腐蚀机理示意图[37−40]Fig.8 Schematic diagram of corrosion mechanism of magnesium[37−40]
当电压低于点蚀电位时,镁合金整个表面覆盖完整的保护膜并且合金的腐蚀速率很低。此时主要是阴极析氢反应:

且析氢速率随着极化电压的升高而降低,直到到达镁合金的点蚀电位。
到达点蚀电位时,镁合金表面膜由于点蚀的作用开始脱落,在镁合金基体裸露区,下列产生单价镁的腐蚀反应发生:

单价的镁离子与水反应生成更为稳定的二价镁的腐蚀产物,此时阳极析氢反应也随之发生:
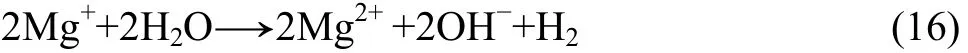
随着电极电位的正移,镁阳极溶解与“阳极析氢”的速度增大,同时由于表面膜的破坏更严重,裸露的金属面积增大,更多镁基体的溶解也使阴极析氢更容易。因此总的析氢速度(为阳极析氢与阴极析氢的总和)就会随着电位的正移而增大,形成负差数效应,而且在腐蚀后期,由于严重的局部或不均匀腐蚀,导致一些未腐蚀的镁颗粒脱落,负差数效应更明显。解释镁合金负差数效应的腐蚀模型的要点可总结为以下几个阶段:
1) 镁阳极材料具有很负的点蚀电位(见图 7),当极化电位达到点蚀电位并继续正移,导致镁合金表面保护膜的破坏;
2) 单价镁离子的溶解以及膜破坏处的阴阳极析氢反应(反应式(13)~(15))使得总的析氢腐蚀速率增加;
3) 腐蚀后期,严重的局部或不均匀腐蚀导致镁颗粒的不均匀脱落。
在此基础上,LIU等[41]根据 Tafel动力学公式对镁阳极的负差数效应用数学模型进行了解释。
4 合金元素和第二相对镁阳极电化学性能的影响
4.1 合金元素
理论上纯镁的电极电位很负,为−2.363 V,但实际应用中,纯镁在水中会产生保护性强的氢氧化镁膜,使材料快速极化[42],因此纯镁不适合做大功率海水电池用阳极。化学成分,即合金元素的种类和含量是影响镁阳极性能最主要的因素。在已有的镁合金阳极中,常用的活化元素主要是Hg、Ga、Sn、Pb、Zn、Mn、Al和Tl等,这些元素都在一定程度上使镁合金的电位负移,改善阳极的电化学活性。
1) 合金元素Hg
研究表明,Hg能通过溶解-再沉积破坏镁表面的钝化膜,维持阳极材料的活化溶解[43]。合金元素 Hg还能有效地抑制镁合金的析氢腐蚀,这与其具有较高的析氢过电位有关。但当Hg的添加量过高时,Hg与Mg形成第二相,易在晶界处析出共晶组织,加速镁阳极的自腐蚀。
2) 合金元素Ga
Ga能与其他合金元素,如Hg、Sn、Pb等,在电极工作温度(60~100 ℃)下形成低共熔混合物,破坏镁表面钝化膜。在含Hg的电解液中,由于Hg和Ga的共同沉积(沉积在镁氧化膜缺陷部位),对镁阳极产生活化作用。必须指出的是,随着 Ga含量的增加,镁合金阳极的电位变负,但当添加量过高时,Ga与Mg形成第二相,在晶界处析出共晶组织,将明显降低电流效率[44]。
3) 合金元素Sn
合金元素 Sn具有较高的析氢过电位,能有效地抑制析氢腐蚀,并能与Ga、In等其他合金元素形成低共熔混合物,破坏Mg表面的钝化膜。Sn还能与Mg形成Mg2Sn相,有利于钝化膜的破裂,使电极反应深入,但同时降低镁合金的耐腐蚀性能[8]。
4) 合金元素Pb
合金元素Pb能与Ga元素形成低共熔混合物,破坏镁表面的钝化膜。Pb的电极电位较Mg正,在电解液中形成微观腐蚀原电池,使镁阳极的电位向正方向漂移。Pb与Al一起加入,降低镁的加工性能,但可改善镁合金阳极的有效工作电位和阳极的电流效率,如AP65合金[45]。
5) 合金元素Zn
Zn 和其他合金元素(Sn、In、Hg、Bi)[45−46]等一起能有效地降低纯镁表面氧化膜的稳定性,从而获得阳极的高活性。Zn能强化镁基体,但对铸造性能有不利影响,有形成疏松和热裂纹的倾向。Zn还可提高含Cu、Ni杂质的镁阳极的稳定性,有利于提高阳极的电流效率,但过高则反而会使电流效率降低。一般而言,Zn的质量分数应低于4%[47]。
6) 合金元素Mn
Mn在镁中的溶解度为3.4%,如果熔炼方法控制得当,可得到含有少量Mn晶体的单相固溶体组织。Mn在镁中主要是净化合金组织,消除杂质元素Fe的影响,减小腐蚀速率,从而提高电流效率。Mn的另一个作用是使 Mg-Mn阳极表面形成比氢氧化镁膜更具有保护作用的水化二氧化锰膜,使析氢作用进一步减弱[48]。最近,有人将少量Ca添加到Mg-Mn合金中,Ca使Mg-Mn合金晶粒细化,并在镁基体的晶界上析出Mg2Ca阴极性化合物,使得晶间腐蚀倾向降低,减少晶粒的脱落,从而提高电流效率,达到62.36%,而且驱动电压也有所增大[48−49]。
7) 合金元素Al
Al元素的加入能使镁阳极表面生成可导电的氢氧化物膜以及细颗粒的反应产物,并迅速从镁阳极表面剥落,有助于提高镁阳极的电流效率[50]。控制 Al的最大含量也至关重要,因为过量的Al容易与Mg反应生成具有阴极特性的中间产物Mg2Al3,这种中间产物会导致晶间腐蚀的发生,降低电流效率,因此镁阳极中的Al含量不宜过高。Al还能提高镁合金强度[47]。早期使用的 Mg阳极材料以及现今使用的大部分 Mg牺牲阳极材料均采用 AZ系列的镁合金,如 AZ31、AZ61和AZ91。
8) 合金元素Tl
Tl元素与Hg、Pb等元素一样属于具有高析氢过电位的重金属元素,Tl能破坏镁阳极表面的保护膜,减少腐蚀产物成泥,降低阳极极化,提高工作电位。英国国防部开发使用的MTA 75(Mg-7%Tl-5%Al)阳极具有比AP65合金更高的工作电位[51]。
4.2 第二相
对于不同的阳极材料,必须考虑第二相粒子的类别、特性等对其电化学性能的影响。冯艳等[16]研究了不同的第二相粒子 Mg3Hg、Mg5Ga2、Mg21Ga5Hg3对Mg-Hg-Ga阳极材料电化学性能的影响,指出这3个第二相对Mg-Hg-Ga阳极的电化学活性的促进作用从高到低依次为Mg3Hg、Mg21Ga5Hg3、Mg5Ga2,交流阻抗分析表明第二相Mg3Hg使Mg-Hg-Ga合金具有最小的法拉第反应电荷传递阻抗 Rct和最大的界面双电层电容Cdlt,该合金电荷传递阻力最小,电极反应迅速,电化学活性最好。含 Mg5Ga2相的 Mg-Hg-Ga阳极具有最大的韦伯阻抗,此合金电极反应中反应物和产物的扩散系数最小,这与合金具有最大的电极表面腐蚀产物电阻 Rox有关,大量腐蚀产物的堆积阻碍电极反应的反应物和产物在电极表面的扩散,导致最大的浓差极化,因此合金的电化学活性降低。
第二相粒子的尺寸、形态和分布对镁阳极的电流效率和表面溶解状态也有比较明显的影响,大小均匀、形态规则、数量适中的第二相粒子使镁阳极表现出较好的电化学性能[52]。
若第二相粒子体积增大,则与镁基体的接触面积也相应增加,第二相作为阴极相导致其周围镁基体优先溶解,贡献的阳极电流和时间持续增加,使得第二相粒子脱溶引起的相对电流效率损失减小,因而阳极电流效率增加,但尺寸较大的第二相粒子将加速阳极的局部溶解,使阳极表面溶解的均匀性降低。
若第二相粒子在晶界连续分布,则引起晶界的局部活化、优先溶解,使阳极保持较低的电流效率和较差的表面溶解均匀性。若第二相粒子在晶内弥散分布,则阳极的活化表现为第二相粒子随镁基体溶解—脱落和合金元素的溶解—再沉积,并且随着第二相粒子之间间距增加,阳极活化的主导因素将由第二相粒子随镁基体的溶解—脱落逐渐转变为合金元素的溶解—再沉积。两者对阳极电流效率的贡献大体相当,因而阳极电流效率维持基本不变,在这种情况下,只要基体中固溶的合金元素能保持足够的活性,都将得到较均匀的表面溶解状态。
若第二相粒子形状规则,则第二相粒子促进镁基体溶解的速度在各个方向上大体相等,因而优先溶解较为充分,第二相粒子脱落引起的阳极电流效率损失相对较小;若第二相粒子形状很不规则,则因其与镁基体的相互镶嵌不容易脱落,阳极电流效率损失也小,因此第二相粒子由规则到不规则,阳极电流效率先下降而后升高。此外,第二相粒子越规则,其周围镁基体的溶解和自身脱溶越均匀,阳极表面的宏观溶解必然也均匀。
冯艳等[8]通过研究第二相 Mg21Ga5Hg3和 Mg5Ga2颗粒的大小、形态和分布对Mg-Hg-Ga阳极电化学性能的影响,指出尺寸为10~20 nm,晶内弥散分布的规则球状Mg21Ga5Hg3颗粒(见图9(a))使Mg-Hg-Ga阳极具有最佳的电化学活性和耐腐蚀性能,而晶内析出的长条状、块状Mg5Ga2第二相(见图9(b))使Mg-Hg-Ga阳极电化学活性未得到明显提高,而且耐腐蚀性能大大降低。

图9 Mg-Hg-Ga阳极晶内析出相的形貌Fig.9 Morphologies of transgranular precipitates in Mg-Hg-Ga anodes
5 结语
镁具有自然资源丰富、物理化学性能良好以及安全性能好的特点,是一种很有发展前景的高能量密度的电池阳极材料。近年来开发了适用于不同功率海水电池各种新型的镁合金阳极,然而海水电池用镁阳极的众多研究领域仍缺乏深入的理论探讨,如镁电极在实际电池体系中的电化学过程动力学特征,镁电极表面微观电化学过程,电解液中离子的运动,众多合金元素,如Zn、Bi、Pb、In的作用机理等。这些研究的不深入导致应用领域的有些问题难以从根本上得到解决,如镁阳极效率的提高,激活状态的改变以及析氢腐蚀的降低等。今后海水电池用镁阳极的发展应在充分研究不同合金元素对镁阳极的活化机理及其电化学动力学过程的基础上,寻找阳极利用率高的镁合金阳极材料,进一步减小析氢的腐蚀,解决活化与钝化的矛盾,提高其比能量。
REFERENCES
[1] 宋玉苏, 王树宗. 海水电池研究及应用[J]. 鱼雷技术, 2004,12(2): 4−8.SONG Yu-su, WANG Shu-zong. Research and application of seawater battery[J]. Torpedo Technology, 2004, 12(2): 4−8.
[2] ZHAO H Y, BIAN P, JU D Y. Electrochemical performance of magnesium alloy and its application on the sea water battery[J].Journal of Environmental Science Supplement, 2009, 21(1):S88−S91.
[3] 王树宗. 鱼雷动力电池技术发展水平概述[J]. 海军工程学院学报, 1994(1): 95−105.WANG Shu-zong. Summary of development of torpedo propulsion technique[J]. Journal of Naval Academy of Engineering, 1994(1): 95−105.
[4] 蔡年生. 国外鱼雷动力电池的发展及应用[J]. 鱼雷技术, 2003,11(1): 12−16.CAI Nian-sheng. Development and application of batteries for overseas torpedo propulsion[J]. Torpedo Technology, 2003, 11(1):12−16.
[5] FONT S, DESCROIX J P, SARRE G. Advanced reserve batteries for torpedoes propulsion[C]//Cherry H. Proceedings of the 31st Power Sources Symposium. Penniton: Electrochemical Society,1984: 362−368.
[6] LI Q F, BJERRUM N J. Aluminum as anode for energy storage and conversion: a review[J]. Journal of Power Sources, 2002,110(1): 1−10.
[7] 马正青, 左 列, 庞 旭, 曾苏民. 铝电池研究进展[J]. 船电技术, 2008, 28(5): 257−261.MA Zheng-qing, ZUO Lie, PANG Xu, ZENG Su-min. Advance in aluminum batteries[J]. Marine Electric and Electronic Technology, 2008, 28(5): 257−261.
[8] 冯 艳. Mg-Hg-Ga阳极材料合金设计及性能优化[D]. 长沙:中南大学, 2009.FENG Yan. Alloy design and properties optimization of Mg-Hg-Ga anode materials[D]. Changsha: Central South University, 2009.
[9] 彭成红, 朱 敏. 镁电池研究进展[J]. 电池, 2003, 33(2):121−123.PENG Cheng-hong, ZHU Min. Development of magnesium batteries[J]. Battery Bimonthly, 2003, 33(2): 121−123.
[10] YAMAMOTO O, KANBARA T, ITO S. Magnesium alloy battery: US 6265109[P]. 2001−07−24.
[11] HIROI M. Pressure effects on the performance and the e.m.f. of the Mg-AgCl seawater battery[J]. Journal of Applied Electrochemistry, 1980, 10(2): 203−211.
[12] 马素卿, 陈亚昕. 水下推进用高能电池[J]. 船电技术, 1999,19(1): 13−23.MA Su-qing, CHEN Ya-xin. High-energy battery for underwater propulsion[J]. Marine Electric & Electronic Technology, 1999,19(1): 13−23.
[13] 奚碚华, 夏 天. 鱼雷动力电池研究进展[J]. 鱼雷技术, 2005,13(2): 7−12.XI Bei-hua, XIA Tian. Survey of power battery for torpedo propulsion[J]. Torpedo Technology, 2005, 13(2): 7−12.
[14] 姜忆初. 电动鱼雷用动力电源及其发展方向[J]. 船电技术,2005, 25(5): 46−48.JIANG Yi-chu. Electric power sources used in electric torpedo and its development trends[J]. Marine Electric and Electronic Technology, 2005, 25(5): 46−48.
[15] 王树宗. A244/s鱼雷动力电池材料与制造工艺分析[J]. 鱼雷技术, 1994, 2(1) : 46−53.WANG Shu-zong. Analysis for manufacture process of A244/s torpedo power battery materials[J]. Torpedo Technology, 1994,2(1): 46−53.
[16] FENG Y, WANG R C, YU K, PENG C Q, ZHANG J P, ZHANG C. Activation of Mg-Hg anodes by Ga in NaCl solution[J].Journal of Alloys and compounds, 2009, 473(1/2): 215−219.
[17] 邓姝皓, 易丹青, 赵丽红, 周玲伶, 王 斌, 冀成年, 兰 博.一种新型海水电池用镁负极材料的研究[J]. 电源技术, 2007,31(5): 402−405.DENG Su-hao, YI Dan-qing, ZHAO Li-hong, ZHOU Ling-ling,WANG Bin, JI Cheng-nian, LAN Bo. Study on Mg alloy anode material for seawater battery[J]. Battery Technology, 2007, 31(5):402−405.
[18] CAO F H, LEN V H, ZHANG Z, ZHANG J Q. Corrosion behavior of magnesium and its alloy in NaCl solution[J].Russian Journal of Electrochemistry, 2007, 43(7): 837−843.
[19] UHLENHAUT D I, FURRER A, UGGOWITZER P J,LOFFLER J F. Corrosion properties of glassy Mg70Al15Ga15 in 0.1 M NaCl solution[J]. Intermetallics, 2009, 17(10): 811−817.
[20] WANG Lei, ZHANG Bo-ping, TADASHI S. Corrosion behavior of AZ91 magnesium alloy in dilute NaCl solutions[J]. Materials and Design, 2010, 31(2): 857−863.
[21] 杨 武. 金属的局部腐蚀−点腐蚀·缝隙腐蚀·晶间腐蚀·成分选择性腐蚀[M]. 北京: 化工出版社, 1995.YANG Wu. Local corrosion of metal pitting corrosion· crevice corrosion· intergranular corrosion· composition selective corrosion[M]. Beijing: Chemical Industry Press, 1995.
[22] 侯军才. 高电位镁阳极的制备及性能研究[D]. 郑州: 郑州大学, 2006.HOU Jun-cai. Preparation and performance research of high-potential magnesium anode[D]. Zhenzhou: University of Zhenzhou, 2006.
[23] 余 琨, 黎文献, 李松瑞. 变形镁合金材料研究进展[J]. 轻合金加工技术, 2001, 29(7): 6−10.YU Kun, LI Wen-xian, LI Song-rui. The research and developments of wrought magnesium alloy[J]. Light Alloy Fabrication Technology, 2001, 29(7): 6−10.
[24] BENDER S, GOELLNER J, ATRENS A. Corrosion of AZ91 in 1 M NaCl and the mechanism of magnesium corrosion[J].Advanced Engineering Materials, 2008, 10(6): 583−587.
[25] PARDO A, MERINO M C, COY A E, ARRABAL R, VIEJO F,MATYKINA F. Corrosion behaviour of magnesium/aluminium alloys in 3.5 wt.% NaCl[J]. Corrosion Science, 2008, 50(3):823−834.
[26] HANAWALT J D, NELSON C E, PELOUBET J A. Corrosion studies of magnesium and its alloys[J]. Transaction of American Institute of Mining, Metallurgical, and Petroleum Engineers,1942, 147: 273−299.
[27] FENG Y, WANG R C, YU K, LI W X. Influence of heat treatment on electrochemical behavior of Mg anode materials[J].Journal of Central South University of Technology, 2007, 2(14):12−15.
[28] SONG G L, ATRENS A. Corrosion mechanisms of magnesium alloys[J]. Advanced Engineering Materials, 1999, 1(1): 11−33.
[29] MAKAR G L, KRUGER K. Corrosion of magnesium[J].International Materials Reviews, 1993, 38(3): 138−153.
[30] MAKAR G L, KRUGER K. Corrosion studies of rapidly solidified magnesium alloys[J]. Journal of Electrochemical Society, 1990, 137(2): 414−421.
[31] UHLIG H H, REVIE R W. Corrosion and corrosion control[M].3rd ed. New York: WILEY J, 1985.
[32] SONG G, ATRENS A, STJOHN D, NAIRN J, LI Y. The electrochemical corrosion of pure magnesium in 1 M NaCl[J].Corrosion Science, 1997, 39(5): 855−875.
[33] LUNDER O, NISANCIOGLU K, HANSEN R S. Corrosion of die cast magnesium-aluminum alloys, SAE Technical Paper Series #930755[R]. Detroit, 1993.
[34] HOEY G R, COHEN M. Corrosion of anodically and cathodically polarized magnesium in aqueous media[J]. Journal of Electrochemical Society, 1958, 105(5): 245−250.
[35] SONG G L, ATRENS A, WU X, BO Z, ZHANG B. Corrosion behaviour of AZ21, AZ501 and AZ91 in sodium chloride[J].Corrosion Science, 1998, 40(10): 1769−1791.
[36] SONG G L, ATRENS A, DARGUSCH M. Influence of microstructure on the corrosion of diecast AZ91D[J]. Corrosion Science, 1999, 41(2): 249−273.
[37] SONG G L, ATRENS A. Recent progress in corrosion and protection of magnesium alloys-An over of cast’s research work[J]. Advanced Engineering Materials, 2005, 7(7): 563−586.[38] SONG G L, ATRENS A. Understanding magnesium corrosion-a framework for improved alloy performance[J]. Advanced Engineering Materials, 2003, 5(12): 837−858.
[39] SONG G L. Investigation on corrosion of magnesium and its alloys[J]. The Journal of Corrosion Science and Engineering,2007, 6: C104.
[40] 宋光铃. 镁合金腐蚀与防护[M]. 北京: 化学工业出版社,2006.SONG Guang-ling. Corrosion and protection of magnesium[M].Beijing: Chemical Industry Press, 2006.
[41] LIU L J, SCHLESINGER M. Corrosion of magnesium and its alloys[J]. Corrosion Science, 2009, 51(8): 1733−1737.
[42] 袁华堂, 吴 峰, 武绪丽, 李 强. 可充镁电池的研究和发展趋势[J]. 电池, 2002, 32(Z1): 14−17.YUAN Hua-tang, WU Feng, WU Xu-li, LI Qiang. The study and development of rechargeable magnesium battery[J]. Battery Nimonthly, 2002, 32(Z1): 14−17.
[43] FENG Yan, WANG Ri-chu, YU Kun, PENG Chao-qun, LI Wen-xian. Influence of Ga and Hg on microstructure and electrochemical corrosion behavior of Mg alloy anode materials[J]. Transactions of Nonferrous Metals Society of China,2007, 17(6): 1363−1366.
[44] FENG Y, WANG R C, YU K, PENG C Q, LI W X. Influence of Ga content on electrochemical behavior of Mg-5at% Hg anode materials[J]. Materials Transactions, 2008, 49(5): 1077−1080.
[45] SAIDMAN S B, BESSONE J B. Cathodic Polarization characteristics and activation of aluminum in chlorid solution containing indium and zinc ions[J]. Journal of Applied Electrochemistry, 1997, 27(6): 731−737.
[46] SHAYEB H A, WAHAB F M A, ABEDIN S Z. Electrochemical behaviour of Al, Al-Sn, Al-Zn, Al-Sn-Zn alloys in chloride solution containing stannousions[J]. Corrosion Science, 2001,43(4): 655−669.
[47] 候德龙, 宋月清, 王 译, 李德富, 何德山. 高电位镁合金(Mg-Mn)阳极熔体净化技术的研究[J]. 稀有金属, 2006, 30(1):30−33.HOU De-long, SONG Yue-qing, WANG Yi, LI De-fu, HE De-shan. Purification technology of Mg-Mn alloy sacrificial anodes[J]. Rare Metal, 2006, 30(1): 30−33.
[48] JUNG G K, JIN H J, SE J K. Development of high-driving potential and high-efficiency Mg-based sacrificial anodes for cathodic protection[J]. Journal of Materials Science Letters,2000, 19(6): 477−479.
[49] JUNG G K, KIM Y W. Advanced Mg-Mn-Ca sacrificial anode materials for cathodic protection[J]. Materials and Corrosion,2001, 52(2): 137−139.
[50] BELDJOUDI T, FIAUD C, ROBBIOLA L. Influence of homogenization and artificial aging heat treatments on corrosion behavior of Mg-Al alloys[J]. Corrosion Science, 1993, 49(9):738−745.
[51] KING J F, UNSWORTH W. Magnesium in seawater batteries[J].Light Metal Age, 1978, 36(7/8): 22−24.
[52] ANIK M, AVCI P, TANVERDI A, CELIKYUREK I, BAKSAN B, GURLER R. Effect of the eutectic phase mixture on the anodic behavior of alloy AZ91[J]. Materials and Design, 2006,27(5): 347−355.
Researches and applications of magnesium anode materials in seawater battery
FENG Yan, WANG Ri-chu, PENG Chao-qun
(School of Materials Science and Engineering, Central South University, Changsha 410083, China)
The magnesium anodes have great development in seawater battery due to their excellent properties, such as good electro negativity, high specific capacity and low density. The researches and applications of magnesium anodes in seawater battery in recent years were reviewed. The influences of activation mechanism, corrosion behavior, alloying elements and second phases on the electrochemical and corrosion properties of magnesium anodes were discussed. It is indicated that the development of magnesium anodes in seawater battery is to prepare novel anode materials with less self-corrosion, higher anode utilization ratio and larger specific energy based on studying activation mechanism of alloying elements and second phases.
seawater battery; magnesium anode; activation; corrosion; alloying element; second phase
TG174.451
A
1004-0609(2011)02-0259-10
国防科工委民口配套研制项目(MKPT-02-181)
2009-12-01;
2010-03-16
王日初,教授,博士;电话:0731-88836638;E-mail:wrc910103@163.com
(编辑 李艳红)
