非晶态石英的变电荷分子动力学模拟*
2011-09-28马颖
马 颖
(湘潭大学材料与光电物理学院,低维材料及其应用技术教育部重点实验室,湘潭 411105)
非晶态石英的变电荷分子动力学模拟*
马 颖
(湘潭大学材料与光电物理学院,低维材料及其应用技术教育部重点实验室,湘潭 411105)
(2010年1月13日收到;2010年4月15日收到修改稿)
基于迭代变电荷方法,用分子动力学模拟了非晶态石英的结构与振动特征.首先利用熔化-猝火方法得到了非晶石英的平衡结构.在此基础上获得了体系不同原子对之间的对关联函数、键角分布函数和振动频谱等,结果与实验数据均符合较好.变电荷方法的计算结果表明,非晶石英体系内粒子的电荷与石英晶体内粒子电荷显著不同,并且出现了较大的涨落.
分子动力学,变电荷,非晶石英
PACS:61.43.Fs,61.43.Bn,63.50.Lm
1.引 言
石英(SiO2)也许是人类历史上认识最早、应用最广泛的材料之一.常温常压下的石英晶体,即 α-石英,属于三方晶系,在现代工业中具有广泛的用途.随着温度、压强等条件的变化,石英还能存在于多种不同的晶体结构中.非晶态石英,俗称玻璃,同样也是重要的工业材料,具有优良的光学、机械、热稳定性能和良好的抗辐照性能,广泛用于微电子、化工、光学等领域[1].因此,人们从实验和理论两方面对晶态和非晶态石英展开了大量的研究,从微观机制上理解其宏观性质[2—5].
近年来,随着计算机软硬件的飞速发展,计算机模拟的手段日益成为材料科学研究中的重要工具.现有的实验手段还很难得到材料在原子尺度上的直观图像,而计算机模拟的方法则无疑能有效地弥补这一缺陷.广泛采用的模拟方法包括第一性原理计算、分子动力学模拟和蒙特卡罗方法等.例如,Liu等[6]运用第一性原理方法计算了各种SiO2晶体的稳定性.与第一性原理方法相比,分子动力学不但能够提供材料在原子尺度上的结构信息,而且由于不考虑体系的电子结构,计算效率要高得多,因此也被广泛地用于研究晶态和非晶态石英.例如,分子动力学模拟表明,石英晶体的 α—β相变为有序—无序型相变[7].非晶态石英的结构、表面、振动谱等也能由分子动力学模拟描述[8—10].
遗憾的是,目前对晶态或非晶态石英的分子动力学模拟大都采用了所谓的固定电荷假设,亦即模拟体系内粒子所带的电荷在模拟过程中保持不变,且同种粒子所带电荷相同.然而,理论和实验证据都表明,粒子所带电荷与粒子所处的环境密切相关[11].如果所模拟的体系为晶体,那么在通常条件下,晶体内原子在平衡位置附近做近简谐振动.因此,原子周围环境基本保持不变,且同类原子所处环境基本相同,固定电荷假设仍然适用[12].对于非晶态物质而言,体系内不再存在长程有序性,因此不同位置的同类原子所处的环境不再相同.在模拟过程中,原子所处的环境也有可能发生较大的改变.可见,对于非晶体系,固定电荷假设不再适用.此外,对于缺陷、表面、界面等非周期性体系,固定电荷假设同样不再适用[13].为了模拟上述体系,人们发展了包括电荷平衡(charge equilibration,Qeq)法[14]、电荷涨落(fluctuation charge,FQ)法[15]、迭代变电荷 (iterative fluctuation charge,IFC)法[16]等的变电荷分子动力学方法.这些方法已经在对分子团[17]、晶界[18]等非周期性体系的分子动力学模拟中得到了成功的应用.对非晶石英的变电荷分子动力学的系统研究则尚未见报道.本文利用迭代变电荷方法对非晶态石英进行了分子动力学模拟.模拟得到的非晶态石英的结构性质等与实验结果符合较好.特别是对体系粒子电荷的分析表明,在非晶态石英中粒子电荷与晶态石英显著不同,且在模拟过程中粒子电荷有较大的涨落.
2.模拟方法
分子动力学模拟的关键在于势函数.对SiO2体系,较早采用的势函数包括 Born-Mayer-Huggins (BMH)势和修正的BMH势等.它们都能较好地模拟非晶石英的结构特征[8].在 BMH势和修正的BMH势中,Si原子和O原子的电荷分别取为+4和-2,亦即认为 Si—O键是完全离子性的.事实上,Si—O键还有部分共价性.第一性原理计算表明,在原子团中,每个 Si—O键之间的电荷转移大约为0.7 e[19].由于Si和O的配位数分别为4和2,对应的离子电荷则分别为 +2.8和 -1.4.当然,在晶态或非晶态SiO2块体材料中,Si和O的电荷与在原子团中的电荷不同.此外,在分子动力学模拟中,往往还适当调节Si和O的电荷以得到最佳的模拟结果.一般而言,在块体(晶态或非晶态)SiO2中,Si和O的电荷分别位于 +1.4—+2.8和 -0.7—-1.4之间.比如 Tsuneyuki等[19]提出的 TTAM势,van Beest等[20]提出的BKS势等,都属于这一类的势函数.然而,BMH势,TTAM势和BKS势等都有一个明显的缺陷,就是将带电离子作为点电荷来处理,利用点电荷间的库仑定律直接计算二者的相互作用.事实上,由量子力学基本原理可知,用“电子云”来描述电子绕核运动更为合适;而两带电离子间的相互作用,则应该计算对应的电子云密度的库仑积分[17].这显然会导致计算量的大幅增加.最近,Ma和 Garofalini[7]提出了一种新的势函数,利用经验公式来计算库仑积分,从而较好地解决了这一问题,并且分子动力学模拟结果表明,该势函数能更精确地给出SiO2的晶体结构、弹性常数和声子色散谱,并成功地模拟了石英晶体的 α—β相变.因此,在本文中,我们采用了 Ma和 Garofalini[7]提出的势函数,该势函数的具体形式及参数详见文献[7].此外,对非晶态材料的分子动力学模拟,有必要采用变电荷方法.在本文中,我们采用了所谓的迭代变电荷方法来得到随环境而变化的离子电荷[16].比之于其他变电荷方法,该方法有计算效率高、程序简便等优点,详见文献[16].为了得到非晶态石英的平衡结构,我们采取了常用的熔化-淬火法[8].首先,随机生成尺寸为3 nm×3 nm×3 nm,净电荷为零的SiO2体系.体系初始密度为2.2 g/cm3,即石英玻璃密度的实验值.由于初始结构中Si原子和O原子的位置是随机生成,并不能代表非晶态石英的真实结构,因此将体系升温至5000 K,并保持30 ps以保证初始结构成为完全无序的非晶态.随后,以降温速率为500 K/20 ps将体系温度降至300 K.在降温过程中,体系温度由Nose-Hoover热浴控制[21,22].当体系温度达到300 K时,采用了 Parrinello-Rahman的恒压分子动力学方法[23],体系压强维持在 1 atm (101.325 kPa),模拟时间为30 ps,从而得到非晶态石英的最终平衡结构.在整个熔化-淬火模拟过程中时间步长均取为1 fs.
3.结果与讨论
为了验证模拟结果的正确性,我们首先考察了平衡态非晶石英的结构性质.图1给出了在300 K下表征非晶态石英结构的Si—O,O—O,Si—Si对关联函数.很明显可以看出,在非晶态石英中仅存在短程有序性,而长程有序性消失.上述对关联函数中第一峰的位置则分别对应于非晶态石英中 Si—O,O—O,Si—Si的平衡距离,详见表1.表中同时列出了Soules[24],Feuston和Garofalini[8]利用固定电荷方法得到的模拟结果,以及实验结果[25,26].不难看出,本文的模拟结果与实验结果符合最好.
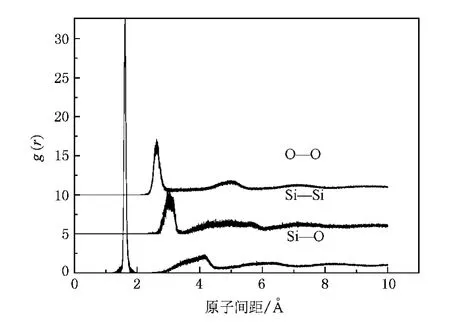
图1 在300 K下表征非晶态石英结构的 Si—O,O—O,Si—Si对关联函数

表1 各种原子间距的比较
除了第一峰位置以外,Si—O—Si键和O—Si—O键的键角分布同样是表征非晶态石英结构的重要参数.图2给出了模拟得到的Si—O—Si键和O—Si—O键键角分布函数.由图可知,O—Si—O键集中分布于100°—120°的区域,峰值位于109°.这一角度与晶态石英中的 O—Si—O键角基本符合,表明在非晶石英中,Si仍然位于四面体中心,而与之相邻的4个O则位于四面体顶点处.Si—O—Si键角的分布则宽泛得多,在110°—180°之间,且无明显峰值.计算可得平均Si—O—Si键角为141°.这些特征与Feuston和Garofalini[8]的固定电荷分子动力学模拟结果基本一致,但平均 Si—O—Si键角比Feuston和 Garofalini的结果小.这是由于二者的电荷分布不同,从而导致了结构上的差异.当然,两种方法的结果均与实验结果基本一致[8].综合对关联函数与键角分布函数的结果可知,非晶态石英实际是由共角的四面体连接而成.在晶态石英中,这些四面体的排布规则,形成了有序的周期性晶体结构.而在非晶石英中,这种有序的周期性消失,但仍然保留了基本的四面体结构单元.
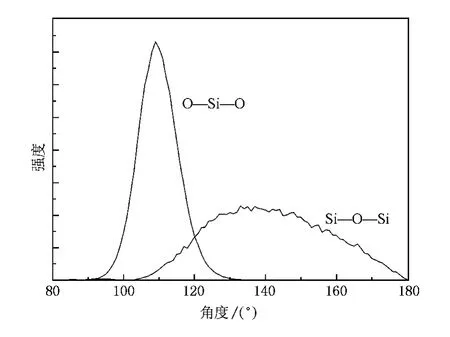
图2 模拟得到的非晶石英中Si—O—Si键和O—Si—O键的键角分布
我们进一步计算了非晶石英的振动频谱.原子的振动频谱直接依赖于原子间的相互作用力.因此,振动频谱的计算可以直接反映分子动力学模拟结果.实验上,非晶态石英的振动谱有三个主要的特征峰,其频率分别为400,800和1100 cm-1,对应于O在垂直于 Si—O—Si平面、在 Si—O—Si平面内沿 Si—O—Si键角角平分线方向和在 Si—O—Si平面内垂直于 Si—O—Si键角角平分线方向的振动.此外,通常还在200 cm-1左右出现一个峰值.在分子动力学中,振动频谱可以通过对速度自关联函数的Fourier变换得到[10].图3给出了非晶石英的振动频谱,其峰值分别位于 187,354,770和 1104 cm-1,均与实验数据符合较好.这无疑进一步证明了本文所采用的势函数和变电荷分子动力学方法的正确性.
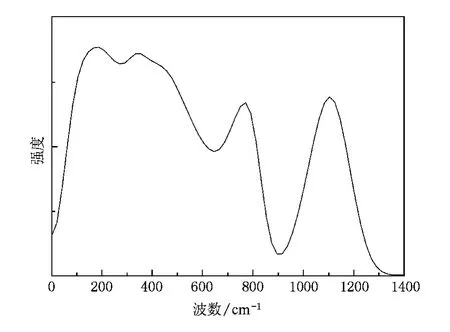
图3 模拟得到的非晶石英的振动频谱
此外,本文采用的变电荷分子动力学方法的计算结果表明,在非晶石英中,Si原子和O原子的电荷均不再是常数,而是出现了较大的涨落.图4和5分别给出了体系内Si原子平均电荷和O原子平均电荷随模拟时间的演化.为便于比较,在图4和5中,我们还给出了用变电荷分子动力学方法得到的相同温度下 α-石英体系内原子平均电荷随模拟时间的演化.由图可知,第一,非晶态与晶态石英内的原子电荷显著不同,这显然是因为二者具有不同的结构.在非晶态石英中,部分Si—O键长仅有1.4左右,对应的 Si原子电荷为1.78,同时部分 Si—O键长增至1.8左右,对应的 Si原子电荷为1.46.而在晶态石英中Si—O键长为1.61,Si原子电荷在1.59左右.这表明随着Si—O键长的减小,Si原子电荷增加较快,而随着Si—O键长的增加,Si原子电荷减小较慢.因此,虽然在非晶石英中的平均Si—O键长仍为1.61,但是Si原子的平均电荷有所增加.类似地,O原子平均电荷则有所降低.原子电荷与键长间的这种非线性关系同样存在于H2O等体系中[27].第二,在同一种体系内,粒子所带电荷随时间而变.这是由于粒子在平衡位置附近振动,对应的其所带电荷也在平衡电荷附近上下涨落.第三,非晶态石英中的粒子电荷涨落较大.这是由于非晶态石英结构较不规则,粒子所处的环境变化较大所致.上述这些特征,显然是任何固定电荷方法所无法描 述 的.虽 然 Feuston 和 Garofalini[8]以 及Garofalini[10]采用的固定电荷分子动力学模拟也能够给出较为精确的非晶石英的结构与振动特征,但是我们的模拟结果表明,变电荷方法还能够正确地描述体系内粒子电荷的变化.
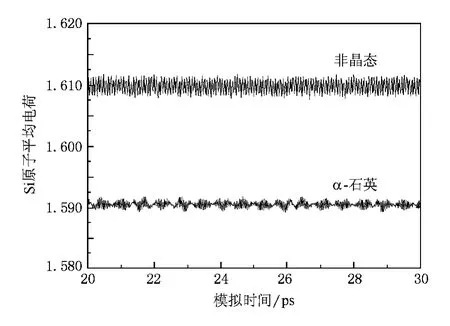
图4 非晶石英和α-石英体系内Si原子平均电荷随模拟时间的演化
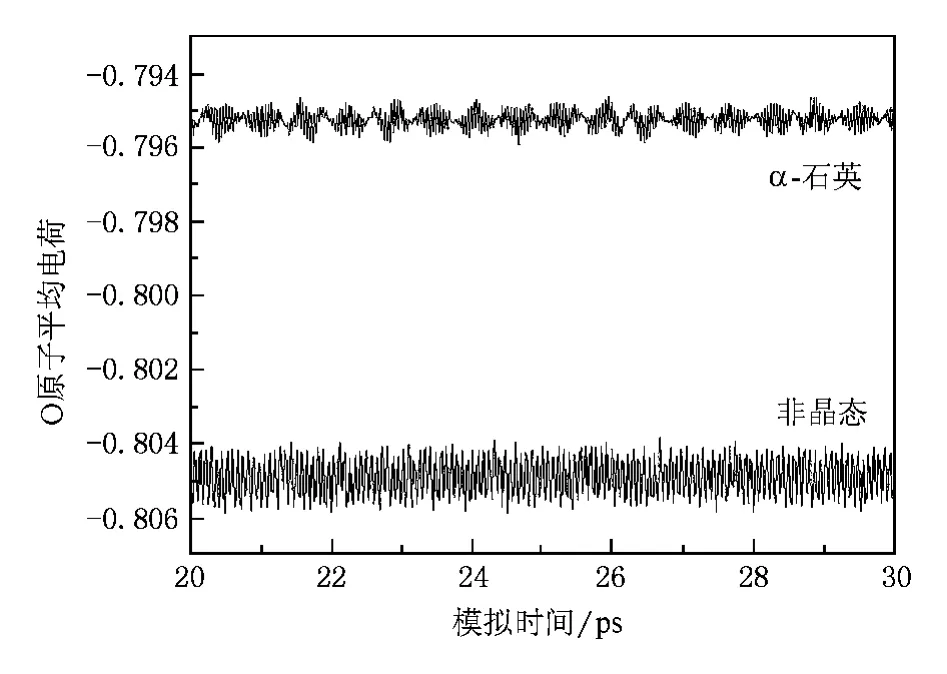
图5 非晶石英和α-石英体系内O原子平均电荷随模拟时间的演化
4.结 论
本文运用变电荷分子动力学方法模拟了非晶态石英的对关联函数、键角分布函数和振动频谱等.此外,还模拟了体系内粒子电荷的演化,这是变电荷方法最显著的特征.在非周期性体系中,这种演化往往显得较为重要.例如,有文献表明SiC中晶界薄膜的电荷转移与薄膜平衡厚度相关[18].本文对非晶态石英的模拟结果表明,变电荷方法不但能够较为精确地描述该体系的结构与振动特性,而且还揭示了非晶石英与晶态石英内原子电荷的不同.本文的成功模拟也表明,本文采用的势函数与变电荷方法还能进一步推广,如用于模拟非晶或晶态石英体系内的缺陷、表面、界面、熔融态石英等非周期性体系.
[1]Zhou Z X,Wang H L,Shen Y Q,Liu D J,Liu H,He S Y,Yang D Z 2008 Acta Phys.Sin.57 592(in Chinese)[周忠祥、王宏利、申艳青、刘大军、刘 海、何世禹、杨德庄2008物理学报57 592]
[2]Yan F P,Wang L,Wei H,Fu Y J,Jian W,Zheng K,Mao X Q,Li J,Liu L S,Peng J,Jian S S 2009 Acta Phys.Sin.58 1793(in Chinese)[延凤平、王 琳、魏 淮、傅永军、简伟、郑 凯、毛向桥、李 坚、刘利松、彭 健、简水生2009物理学报58 1793]
[3]Jia W Y,Pei L W 1984 Acta Phys.Sin.33 1092(in Chinese)[贾惟义、裴力伟1984物理学报33 1092]
[4]Gao S J,Ouyang S X 2003 Acta Phys.Sin.52 1292(in Chinese)[高祀建、欧阳世翕2003物理学报52 1292]
[5]Wang J F,Li H L,Tang R M,Cha J X,He S A 1982 Acta Phys.Sin.31 1423(in Chinese)[王积方、李华丽、唐汝明、查济璇、何寿安1982物理学报31 1423]
[6]Liu F,Garofalini S H,King-Smith D,Vanderbilt D 1993 Phys. Rev.B 49 12528
[7]Ma Y,Garofalini S H 2006 Phys.Rev.B 73 174109
[8]Feuston B P,Garofalini S H 1988 J.Chem.Phys.89 5818
[9]Feuston B P,Garofalini S H 1989 J.Chem.Phys.91 564
[10]Garofalini S H 1982 J.Chem.Phys.76 3189
[11]Mortier W J,Ghosh S K,Shankar S 1986 J.Am.Chem.Soc. 108 4315
[12]Ma Y,Garofalini S H 2008 J.Chem.Phys.128 084505
[13]Nakano A 1997 Comp.Phys.Commun.104 59
[14]Rappe A K,Goddard W A 1991 J.Phys.Chem.95 3358
[15]Rick S W,Stuart S J,Berne B J 1994 J.Chem.Phys.101 6141
[16]Ma Y,Garofalini S H 2006 J.Chem.Phys.124 234102
[17]Louwen J N,Vogt E T C 1998 J.Mol.Catal.A 134 63
[18]Ma Y,Chen S D,Xie G F 2009 Acta Phys.Sin.58 7792(in Chinese)[马 颖、陈尚达、谢国锋2009物理学报58 7792]
[19]Tsuneyuki S,Tsukada M,Aoki H,Matsui Y 1988 Phys.Rev. Lett.61 869
[20]van Beest W H,Kramer G,Santen R A 1990 Phys.Rev.Lett. 64 1955
[21]Nose S 1984 Mol.Phys.52 255
[22]Hoover W G 1985 Phys.Rev.A 31 1695
[23]Parrinello M,Rahman A 1980 Phys.Rev.Lett.45 1196
[24]Soules T F 1979 J.Chem.Phys.71 4570
[25]Mozzi R L,Warren B E 1969 J.Appl.Crystallogr.2 164
[26]Narten A H 1972 J.Chem.Phys.56 1905
[27]Guillot B,Guissani Y 2001 J.Chem.Phys.114 6720
PACS:61.43.Fs,61.43.Bn,63.50.Lm
Variable charge molecular dynamics simulation of vitreous silica*
Ma Ying
(Key Laboratory of Low Dimensional Materials and Application Technology of Ministry of Education,Faculty of Materials,Optoelectronics and Physics,Xiangtan University,Xiangtan 411105,China)
13 January 2010;revised manuscript
15 April 2010)
Variable charge molecular dynamics simulations of vitreous silica have been performed based on the iterative fluctuation charge model.The vitreous silica was formed using the standard melt-quench process.The pair distribution function,angle distribution and frequency spectrum were then obtained.The results are in good agreement with experimental data.More importantly,our results show that the atomic charge in vitreous silica is significantly different from those in the crystalline silica,and larger fluctuations are observed.
molecular dynamics,variable charge,vitreous silica
*国家自然科学基金(批准号:10702059)、高等学校博士学科点专项科研基金 (新教师基金)(批准号:20070530009)、教育部留学回国人员科研启动基金(批准号:2008890)、国家博士后科学基金(批准号:20090451102,201003515)和湘潭大学材料科学与工程博士后流动站资助的课题.
*Project supported by the National Natural Science Foundation of China(Grant No.10702059),the Research Fund for the Youth Scholars of the Specialized Research Fund for the Doctoral Program of Higher Education of China(Grant No.20070530009),the Scientific Research Starting Foundation for the Returned Overseas Chinese Scholars of Ministry of Education,China(Grant No.2008890),the China Postdoctoral Science Foundation(Grant Nos.20090451102,201003515),and the Post-Doctoral Station of Materials Science and Engineering at Xiangtan University,China.
