纳米羟基磷灰石/壳聚糖载药微球的制备及性能
2011-02-06李湘南陈晓明彭志明李世普
李湘南,陈晓明,彭志明,李世普
(1. 武汉理工大学 生物材料与工程研究中心,湖北 武汉,430070;2. 武汉理工大学 化工学院,湖北 武汉,430070)
纳米羟基磷灰石/壳聚糖载药微球的制备及性能
李湘南1,2,陈晓明1,彭志明1,李世普1
(1. 武汉理工大学 生物材料与工程研究中心,湖北 武汉,430070;2. 武汉理工大学 化工学院,湖北 武汉,430070)
以纳米羟基磷灰石和壳聚糖为基质,构建一种新型甲硝唑缓释微球,作为充填材料用于骨修复。利用乙醇为反应溶剂,聚丙烯酸为分散剂,在pH=11的条件下,制备针状纳米羟基磷灰石。采用W/O 型反相乳化−交联技术制备羟基磷灰石/壳聚糖载甲硝唑复合微球。通过紫外分光光度法测定甲硝唑含量和体外累积释放度。研究结果表明:制得的羟基磷灰石/壳聚糖载药复合微球粒径主要集中在1~10 μm,壳聚糖对羟基磷灰石和甲硝唑形成了很好的包覆。复合微球平均载药量为38.23%,平均包封率为54.21%,3 d内对甲硝唑的释放达到82%左右。所制备的羟基磷灰石/壳聚糖载药复合微球形态圆整,粒径分布较为均匀,对甲硝唑具有较好的缓释效果。
缓释微球;甲硝唑;纳米羟基磷灰石;壳聚糖
甲硝唑具有广谱抗厌氧菌和抗原虫的作用,早在1978年就被世界卫生组织选定为抗厌氧菌的基本药物,临床主要用于预防和治疗厌氧菌引起的感染,如呼吸道、消化道、腹腔及盆脓感染,皮肤软组织、骨和骨关节等部位的感染以及脆弱拟杆菌引起的心内膜炎、败血症及脑膜炎等,此外,还广泛应用于预防和治疗口腔厌氧菌感染[1]。羟基磷酸钙是脊椎动物的骨组织的主要无机成分,人工合成羟基磷灰石(HAP)的理化性质、结构与其基本相同,无毒副作用,无刺激性,动物实验和临床观察都证明该材料是一种生物相容性极好的骨代用品,具有骨引导作用,填入骨缺损部位后能为骨基质的沉积和维持提供一个良好的骨床,引导周围骨组织增生,加快成骨过程,为新骨的改建创造了良好条件,从而促进缺损骨的愈合[2−6]。但HAP 降解速度慢,颗粒之间无黏着性,凝固缓慢,无论充填拔牙窝、骨腔,还是用于骨组织支架材料等都会出现HAP颗粒外溢和难以保持外形的情况[7]。壳聚糖(CS)即羧甲基脱乙酰壳多糖来源方便,易提取,是自然界少见的带正电荷的高分子聚合物,无毒性,无刺激性,不致敏,不与体液发生反应并可被机体的溶解酶生物降解,具有良好的生物相容性和独特的生物学活性,并有促进伤口愈合、骨形成等功能,可被人体吸收利用;壳聚糖具有一定黏度,能改善混合物的流动性和剂型,有消炎止痛和促进伤口愈合作用[8−9]。羟基磷灰石与壳聚糖复合作为组织工程材料的报道很多,但多为粉体材料或块状材料[10−13]。有研究表明:颗粒的形态和结构是影响颗粒在人体内活性的主要因素,形状不规则的载药颗粒植入人体会产生炎症反应,因此,形状规则的球状载药颗粒是植入药物载体的首选[14−16]。本文作者用甲硝唑为主药、HAP和壳聚糖为载体制成具有可调药物释放性能的羟基磷灰石/壳聚糖缓释微球作为骨填充材料进行局部定向释药,利用纳米HAP的大比表面积和高表面活性装载抗菌药物,以及壳聚糖对药物的包埋功能起到缓释药物的作用,微球材料能很好地与骨相容为一体,从而使骨缺损部位伤口愈合加快,达到骨修复的目的。
1 实验过程
1.1 实验材料
材料为:壳聚糖粉末,购自浙江玉环县海洋生物化学有限公司;脱乙酰度为92%,平均相对分子质量为8×105,医用级。羟基磷灰石,为实验室自制;其他试剂均为分析纯,购自国药集团。
1.2 纳米羟基磷灰石的制备及表征
将5.93 g Ca(NO3)2·4H2O加50%(体积分数)乙醇溶液150 mL配制得溶液1,将2.0 g (NH4)2HPO4加入50%乙醇溶液150 mL中,配制成溶液2。在常温下,将溶液2滴入溶液1中,强力搅拌(转速为1 000 r/min),并滴加微量聚丙烯酸(PAA),同时以2.0 mol/L的氨水为pH调节剂,调节pH为11.0,滴加完毕后继续搅拌反应3 h(保持室温下进行);得悬浮液,静置陈化12 h,离心后用去离子水和无水乙醇各洗3遍,离心分离后产物在50 ℃真空干燥。
1.3 壳聚糖/HAP复合微球的制备
采用反相乳液法制备壳聚糖/HAP复合微球。在60 mL 2%(质量分数)乙酸水溶液中加入1.5 g壳聚糖粉末,搅拌溶解,按壳聚糖、羟基磷灰石质量比为1:1称取适量羟基磷灰石粉,与壳聚糖溶液通过超声和磁力搅拌混合均匀后,将其倾倒入160 mL液体石蜡中,搅拌30 min后加入5 mL span80高速搅拌乳化,乳化后加入5 mL 25%(质量分数)戊二醛在50 ℃水浴中交联10 h,将得到的悬浮液抽滤,用氯仿和无水乙醇依次各洗3次,在50 ℃真空干燥得到壳聚糖/HAP复合微球。
1.4 载甲硝唑壳聚糖/HAP复合微球的制备
采用反相乳液法,按壳聚糖、羟基磷灰石和甲硝唑质量比为1:1:1投料制备载甲硝唑壳聚糖/HAP复合微球。
1.5 微球表征
分别用X线衍射仪(XRD, D/Max-IIIA, Rigaku, Japan)和透射电镜(TEM, JEM-2100F, JEOL, Japan)对纳米羟基磷灰石样品进行物相分析和形貌观察。
采用扫描电镜(SEM, H-600 STEM/EDX PV9100, HITACHI, Japan)对空白微球和载药微球形貌进行观察;EDS对载药微球载药前后成分变化进行分析;采用KBr压片法制样;采用傅里叶转换红外光谱仪(FT-IR, Nicolet Nexus 670)测定壳聚糖/HAP复合微球的傅里叶转换红外光谱。
1.6 载药量和包封率测定
通过紫外分光光度计(岛津UV2550紫外可见分光仪)测定甲硝唑载药微球的载药量和包封率。
取50 mg甲硝唑对照品,加适量生理盐水溶解后,用生理盐水定容至500 mL。精密吸取0,1.0,2.0,3.0,3.5,4.0,6.0和8.0 mL,分别加生理盐水定容至25 mL,制成0,4,8,12,16,24和32 mg/L的系列标准溶液,以生理盐水为空白样,于200~800 nm范围内扫描,确定甲硝唑最大吸收波长,于最大吸收波长处测定吸光度,回归得到标准曲线。
取微球适量充分研磨,精密称取50 mg用生理盐水定容至100 mL,搅拌24 h后,取其上清液3 mL,过滤后用生理盐水定容于25 mL容量瓶中,在波长为320 nm处测吸光度,根据标准曲线计算出药物质量浓度,进而得微球载药量:
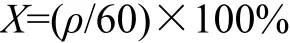
式中:ρ为药物质量浓度,mg/L;X为载药量。
根据微球的质量及微球的载药量,可计算出包封率:
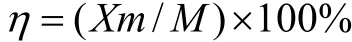
式中:η为包封率;M为投入的总药量;m为微球的质量。
1.7 体外累积释放度测定方法
采用中国药典溶出度测定体外累积释放度。在6个烧杯中各装入 200 mL pH为7.4的磷酸盐缓冲溶液,在37 ℃下以100 r/min搅拌。取微球1.2 g,分别投入6个烧杯中。每隔一定时间分别精密取样2 mL,立即经孔径为0.8 μm微孔滤膜过滤,同时补加缓冲液2 mL。将取出的2 mL置于10 mL容量瓶中,加蒸馏水定容至刻度,摇匀,取空白微球,进行同法操作得对照试样,在波长320 nm下,采用紫外分光光度法分别测定各样品的吸光度,取平均值计算甲硝唑的体外累积释放度。
2 实验结果
2.1 羟基磷灰石的X线衍射分析
羟基磷灰石样品的X线衍射图谱如图1所示。由图1可见,羟基磷灰石的各衍射峰的位置都与标准卡片(JCPDS73-0293)相符,纳米HAP的晶面如(002),(211),(112),(300),(202),(213)和(310)等均已出现特征衍射峰。其中,(002),(211)和(300)晶面的衍射峰为其主要衍射峰,说明最终产物主要为羟基磷灰石弱晶体。
2.2 羟基磷灰石的透射电镜分析
羟基磷灰石粉末的透射电镜照片如图2所示。从图2可以看到,制备的羟基磷灰石粒子为纳米相针棒状晶体颗粒。样品的长径比为3.0~4.0,平均直径及长度分别为5 nm和100 nm左右;纳米颗粒在透射电镜下显示了一定程度的团聚,对于短柱状和针状纳米颗粒,团聚为网络状,粒子之间的接触为点接触式,属于软团聚,不是无法实现再分散的硬团聚,这些颗粒的团聚应该是电镜制样过程中出现的。
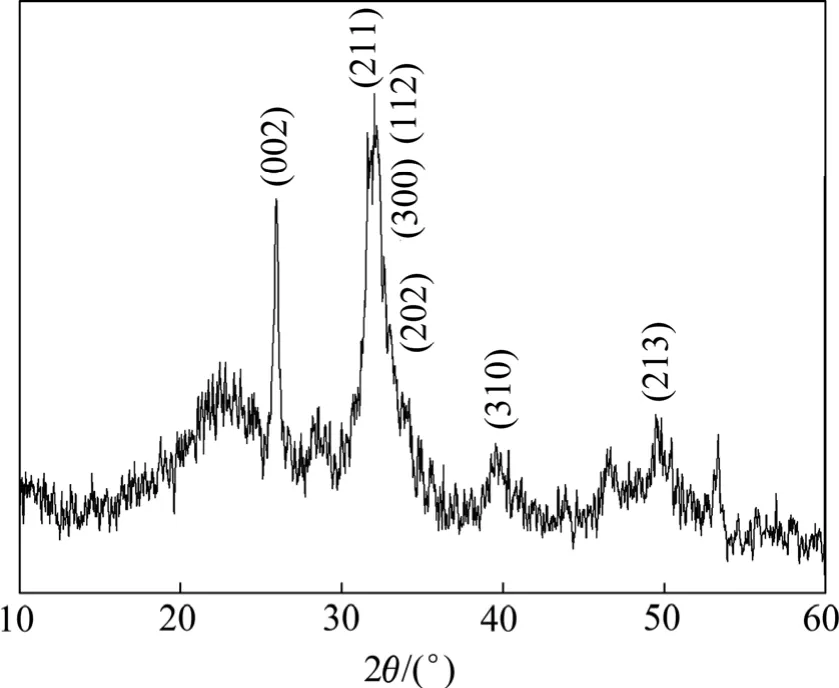
图1 羟基磷灰石样品的X线衍射图谱Fig.1 X-ray diffraction pattern of hydroxyapatite

图2 羟基磷灰石粉末的透射电镜照片Fig.2 TEM image of hydroxyapatite crystals
2.3 微球的形貌
空白微球和载药微球的扫描电镜照片和EPS图谱如图3所示。从图3可见:肉眼观察微球呈小颗粒状,颜色微黄,SEM下观察微球外观较圆整,成球性好。载药前后微球粒径基本没变化,微球粒径分布较均匀,粒径范围为1~10 μm。空白微球表面光滑,载药微球表面粗糙,有少量甲硝唑晶体沉积在微球表面。
从图3(e)可见:C和O波峰较大,而Ca和P波峰较小。这表明空白微球中CS网络结构对HAP形成了良好的包埋,只有极小部分HAP分布在微球表层。从图3(f)可见:出现了弱的N峰,Ca和P峰面积增加,这表明微球中已成功载入了甲硝唑,由于甲硝唑引入的N元素含量较少,在EDS谱图中出现的N峰也较小,同时,载药微球原料配比中壳聚糖的质量分数减小,对HAP的包埋能力下降,使分布在微球表层HAP的含量增加,从而使Ca和P峰面积增加。
2.4 复合微球的红外分析
纯CS,HAP和复合微球的FTIR图谱如图4所示。从图4可见:CS的红外图谱中1 649 cm−1处的吸收峰为酰胺的酰胺Ⅰ带吸收(即C=O的伸缩振动吸收),1 599 cm−1处的肩峰则为胺基的弯曲振动吸收峰(即酰胺Ⅱ谱带),而在3 450 cm−1附近出现的吸收峰归属为O—H及—NH2的N—H伸缩振动吸收峰。
羟基磷灰石的红外图谱中474,571,601,962和1 032~1 087 cm−1处都存在明显的吸收峰,对应于水的吸收带在1 634和3 000~3 700 cm−1处,表征羟基磷灰石生成的OH−吸收峰在630和3 570 cm−1处出现。

图3 微球扫描电镜照片及EDS图谱Fig.3 SEM images and EDS spectra of microspheres
HAP和CS在复合前后没有发生明显变化,在3 450 cm−1处附近的O—H和N—H伸缩峰变宽,并向高波移动,说明复合材料中CS分子与HAP分子间有很强的相互作用。受HAP的影响,复合微球红外图谱中的酰胺Ⅰ带发生了红移现象,1 599 cm−1处的肩峰与酰胺Ⅰ带(1 649 cm−1处)分开,可能是CS的—NH2与HAP中的—OH之间形成了氢键作用或者是细小的HAP颗粒进入了CS的分子链间破坏了链间氢键作用而致。所以,复合微球中同时出现了CS与HAP的特征吸收峰,说明复合微球存在HAP和CS成分。
2.5 载药量和包封率
甲哨唑紫外吸收曲线和数据回归曲线如图5所示。从图5可见:甲硝唑于320 nm处有最大吸收,在320 nm处测定数据进行回归得到标准曲线:y=0.051 68x−0.020 80 (x为质量浓度,mg/L),相关系数r为0.999 9,该曲线线性范围为0~32 mg/L。
制备3批复合微球进行测试,计算得到平均载药量为38.23%,平均包封率为54.21%。
2.6 载药微球的体外释药性能
载药微球的累积释放曲线如图6所示。由图6可知:载药微球在 0~72 h内体外累积释放度逐渐递增,10 h时体外累积释放度为21.02%,35 h时为60%, 55 h时达到80%,随后趋于平缓。这显示本研究所得缓释微球对甲硝唑具有较好的缓释效果,55 h左右时达到最大释放,复合微球在3 d内的体外累积释药分数为0.826 5,释药平稳。
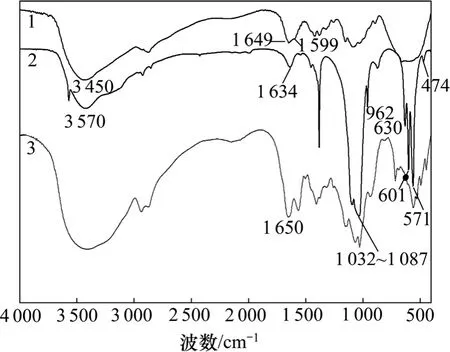
图4 纯CS、纯HAP和复合微球的FTIR图谱Fig.4 Fourier transform infrared spectra of CS, HAP and HAP/CS microspheres
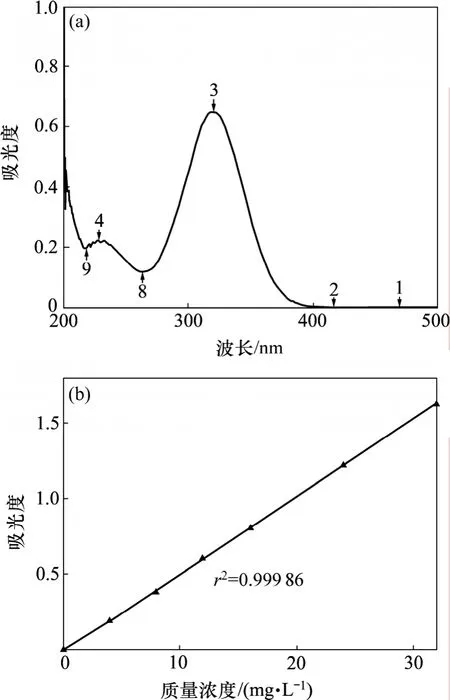
图5 甲硝唑紫外吸收曲线和标准曲线Fig.5 UV spectrum of metronidazole and standard curve
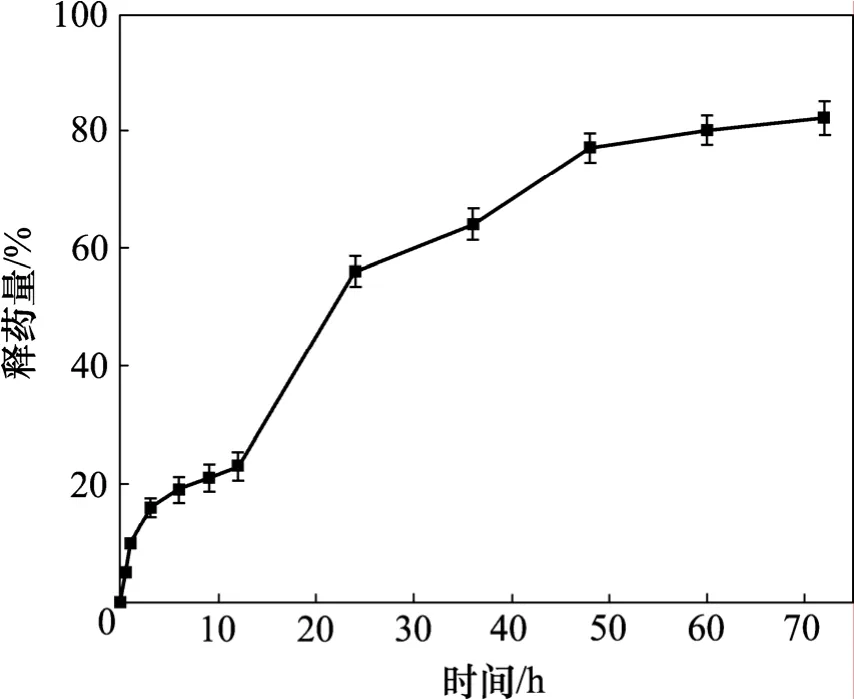
图6 甲硝唑-羟基磷灰石/壳聚糖载药微球的累积释放曲线Fig.6 Cumulative release curve of metronidazole-loaded HAP/CS composite microspheres
3 讨论
本实验采用了反相乳液-交联法,利用纳米羟基磷灰石的巨大表面积和壳聚糖的包埋功能制备了载甲硝唑的复合微球,得到的微球粒径较均一,分散性较好。制得的微球作为骨修复组织工程材料使用时,由于微球的平均直径为8 μm左右,体积较小,可以以注入的方式治疗,是一种较为理想的骨修复材料。
有机−无机微球是近些年骨修复材料及药物新剂型研究的一个热点。这是因为有机−无机复合微球既具有有机材料的可塑性、易加工性以及生物兼容性,又具备无机物的刚性、骨引导活性等性能;同时,可实现对药物的控制释放。
在实验中,用50%乙醇为反应溶剂,当初始溶液pH等于11时,样品都呈针棒状。样品的平均直径及长度分别为30 nm和100 nm,为纳米级别,且分散度较高。在包埋过程中,用超声波加快速搅拌分散在壳聚糖中,避免了羟基磷灰石的团聚。从图3(a)可以看到:微球球形度好,没有看到大块的羟基磷灰石的团聚;从图3(b)可以看到:复合微球表面较为圆整光滑,表面无块状物的团聚,证明羟基磷灰石纳米粉末已均匀分布在壳聚糖网络中,从仿生学角度出发,应当保持骨替代物中羟基磷灰石呈纳米状态,在制备骨修复材料时应尽可能避免羟基磷灰石颗粒发生团聚,本实验结果符合仿生学原理。
在微球中加入甲硝唑,可以预防感染,同时因为药物在高分子载体中,还可以起到缓释的效果。在微球载药量的测定中,0.1 g空白微球可以装载甲硝唑54.21 mg,其载药量为38.23%(质量分数),从图6可知:甲硝唑在72 h内都可以保持有效浓度。
在临床骨创伤的实例中,由于伤口附近的血液循环系统被破坏,常伴随着伤口的感染;为了治疗或预防这种感染,常常要注射一些抗生素药物。很多药物无法直接使用,或者直接使用效果不明显,需要用高分子材料或无机多孔空心来包埋药物,并通过合理地设计微球尺寸、表面性质、缓释性能等来达到在所需的时间、所需的地点以及所需的速度释放出药物。
4 结论
(1) 采用反相乳液−交联法制备的羟基磷灰石/壳聚糖载药微球形态均匀一致,均为圆球状,粒径分布窄,再分散性较好。
(2) 复合微球对甲硝唑的平均载药量为38.23%,平均包封率为54.21%,该载药微球释药平稳,55 h左右时达到最大释放,3 d释药后的甲硝唑体外累积释药分数达到0.826 5。表明该微球对甲硝唑具有较好的载药和缓释性能,可用于进一步骨修复实验研究。
[1] 康现武, 王强, 王毅, 等. 甲硝唑羧甲基壳聚糖缓释微球的制备及体外释放[J]. 药学实践杂志, 2008, 26(3): 182−184.
KANG Xian-wu, WANG Qiang, WANG Yi, et al. Preparation technology and in-vitro release characteristics of metronidazoleloaded carboxymethyl chitosan sustained-release microspheres[J]. Journal of Pharmaceutical Practice, 2008, 26(3): 182−184.
[2] LAI Chen, TANG Shao-qiu, WANG Ying-jun, et al. Formation of calcium phosphate nanoparticles in reverse microemulsions[J]. Mater Lett, 2005, 59(2/3): 210−214.
[3] Kalita S J, Bhardwaj A, Bhatt H A. Nanocrystalline calcium phosphate ceramics in biomedical engineering[J]. Mater Sci Eng C, 2007, 27(3): 441−449.
[4] Domingo C, Arcis R W, Osorio E, et al. Hydrolytic stability of experimental hydroxyapatite-filled dental composite materials[J]. Dent Mater, 2003, 19(6): 478−486.
[5] ZHANG Hai-bin, ZHOU Ke-chao, LI Zhi-you, et al. Synthesis of hollow hybrid hydroxyapatite microspheres based on chitosan-poly(acrylic acid) microparticles[J]. Biomed Mater, 2009, 4(3): 031002.
[6] KONG Xiang-dong, SUN Xiao-dan, LU Jun-biao, et al. Mineralization of calcium phosphate in reverse microemulsion[J]. Current Appl Phys, 2005, 5(5): 519−521.
[7] 石浦江, 李玉宝, 张利, 等. 载微球纳米轻基磷灰石/壳聚糖复合多孔支架的制备与表征[J]. 功能材料, 2006, 37(11): 178−184.
SHI Pu-jiang, LI Yu-bao, ZHANG Li, et al. Fabrication and characterization ofn-HA/CS porous scaffold containing ALG/CS microspheres[J]. Journal of Functional Materials, 2006, 37(11): 178−184.
[8] LU Guang-yuan, ZHU Lin, KONG Li-jun, et al. Porous chitosan microcarriers for large scale cultivation of cells for tissue engineering: Fabrication and evaluation[J]. Tsinghua Sci & Tech, 2006, 11(4): 427−432.
[9] Sinha V R, Singla A K, Wadhawan S, et al. Chitosan microspheresas a potential carrier for drugs[J]. Int J Pharm, 2004, 274(1/2): 1−33.
[10] LIU Hua, LI Hong, CHENG Wen-jun, et al. Novel injectable calcium phosphate/chitosan composites for bone substitute materials[J]. Acta Biomater, 2006, 2(5): 557−565.
[11] HU Qiao-ling, LI Bao-qiang, WANG Mang, et al. Preparation and characterization of biodegradable chitosan/hydroxyapatite nanocomposite rods via in situ hybridization: a potential material as internal fixation of bone fracture[J]. Biomaterials, 2004, 25(5): 779−785.
[12] WANG Li, LI Chun-zhong. Preparation and physicochemical properties of a novel hydroxyapatite/chitosan-silk fibroin composite[J]. Carbohydr Polym, 2007, 68(4): 740−745.
[13] 赵宏霞, 黄才欢, 金花, 等. 壳聚糖/羟基磷灰石−庆大霉素缓释材料的抗菌性能研究及AFM观察[J]. 功能材料, 2009, 40(3): 429−432.
ZHAO Hong-xia, HUANG Cai-huan, JIN Hua, et al. In vitro antibacterial activity evaluations and AFM observations of chitosan/hydroxyapatite-gentamicin as drug delivery system[J]. Journal of Functional Materials, 2009, 40(3): 429−432.
[14] Sivakumarm M, Manjubala I, Rao P K. Preparation, characterization and in-vitro release of gentamicin from coralline hydroxyapatite-chitosan composite microspheres[J]. Carbohydr Polym, 2002, 49(3): 281−288.
[15] Wilson O C, Hull J R. Surface modification of nanophase hydroxyapatite with chitosan[J]. Mater Sci Eng C, 2008, 28(3): 434−437.
[16] 黄大建, 张文熊, 刘晶冰, 等. 纳米羟基磷灰石/壳聚糖−明胶复合微球的制备及性能[J]. 中国组织工程研究与临床康复, 2009, 13(16): 3097−3100.
HUANG Da-jian, ZHANG Wen-xiong, LIU Jing-bing, et al. Preparation and performance of nanohydroxyapatite/ chitosanelatin composite microspheres[J]. Journal of Clinical Rehabilitative Tissue Engineering Research, 2009 13(16): 3097−3100.
(编辑 赵俊)
Preparation and performance of drug-loaded nano-hydroxyapatite/chitosan microspheres
LI Xiang-nan1,2, CHEN Xiao-ming1, PENG Zhi-ming1, LI Shi-pu1
(1. Biomedical Materials and Engineering Research Center, Wuhan University of Technology, Wuhan 430070, China; 2. College of Chemical Engineering, Wuhan University of Technology, Wuhan 430070, China)
Nano-hydroxyapatite and chitosan were selected as matrix to prepare a novel metronidazole sustained release microsphere for bone tissue recovery. Aciform nano-hydroxyapatite was synthesized in ethanol medium (pH=11) using polyacrylic acid as dispersant. Metronidazole-loaded chitosan/hydroxyapatite composite microspheres were prepared by water in oil emulsion cross-linking method. The results show that the size of as-fabricated composite microspheres is mainly distributed from 1 to 10 μm, and hydroxyapatite and metronidazole are embedded very well by chitosan. The average drug loading is 38.23%, the average entrapment efficiency is 54.21%, and the cumulative release rate to metronidazole is near 82% during 3 d. The composite microspheres are spherical and distributed uniformly in diameter with a good property for the slow-release of metronidazole, which have great potential in drug delivery system.
sustained release microsphere; metronidazole; nano-hydroxyapatite; chitosan
R318
A
1672−7207(2011)05−1232−06
2010−01−25;
2010−05−20
国家自然科学基金资助项目(50872099)
李湘南(1977−),女,湖南平江人,博士研究生,从事生物材料研究;电话:15327392976;E-mail: xiangnanlidong@163.com
