偏二甲肼分子化学键解离能的理论计算
2011-01-28尹东光高文亮张彩霞张静静张海东
尹东光,高文亮,张彩霞,张静静,张海东
(1.重庆大学化学化工学院,重庆 400044;2.重庆工商大学废油资源化装备与技术教育部工程研究中心,重庆400067)
引 言
偏二甲肼(UDM H)是一种广泛应用的液体火箭推进剂,同时它又是一种高毒性化合物。李正莉[1]等采用量子化学计算方法对其分子结构和化学反应性能进行了研究。相关手册[2]中收录其化学键解离能的数据相对陈旧,有必要进行严格的理论计算加以比较和验证。
化学键解离能(BDE)是反映物质化学键强弱的一种重要参数[2]。Hartree-Fock(H-F)及微扰理论(M Pn)方法因为在处理开壳层体系存在自旋污染而不适合BDE 的计算,更高级别的从头算法如CCSD、QCISD 等不仅由于计算量大,应用的体系受到限制,而且当采用的基组不够大时仍会大大低估BDE 值。为了克服以上方法的不足,J.A.Pople 和G.A.Petersson 分别发展了G aussian-n(G n)理论和CBS(Complete Basis Sets)理论的组合从头算方法。Gn 理论一般采用M P2(FU)/6-31G(d)进行分子几何构型优化,如G3[3]理论,而G3B3[4]与G3的区别在于其使用密度泛函理论的B3LYP 方法进行几何构型的优化和零点振动能计算。CBS-Q[5]方法采用UM P2(FC)/6-31G+几何优化构型,CBSQB3[6]与CBS-Q 的区别类似于G3B3 和G3。
组合从头算方法可以作为BDE 的标准预测方法[7]。Hongyan Sun 等[8]利用CBS-QB3 方法,通过等键反应得到甲基肼(MM H)中CH 3N(—H)NH 2 键解离能为330.5 kJ/mol,与郭庆祥等[9]利用G3B3 得到的结果320.1 kJ/mol 相差仅为10.4 kJ/mol。M .A.Bohn[10]利 用 B3LYP 方 法 对 肼(N2H4)、甲基肼、偏二甲肼和1,2-二甲肼(SDM H)的N—N 键BDE 进行了计算。
本研究利用量子化学组合从头算法和密度泛函理论方法对UDM H 分子的BDE 正值进行了计算和比较,以期能获得准确的UDM H 的BDE 值。
1 计算模型及方法
除组合从头算方法以外,UDM H 分子及相关自由基的几何构型在(U)B3LYP[11]/6-31G(d)的水平上进行优化,并在同一水平进行振动频率计算,所得频率值均为正值,无虚频存在,证明所得结构均为稳定结构。然后分别采用B3LYP 、BM K[12]和B3P86[13]泛函方法,对偏二甲肼及相关自由基进行单点电子能量的计算,单点能的计算基组采用6-311++G(2df,2p),对焓值的校正项采用(U)B3LYP/6-31G(d)水平的结果。所有计算采用Gaussian 03[14]程序包完成。
2 结果与讨论
2.1 组合从头算方法的计算结果
表1 列出了组合从头算方法对偏二甲肼BDE的计算结果。从表1可以看出,几种组合从头算方法得到的BDE 数据非常接近,相差仅为几个kJ/mol,尤其是对N—N、N—H 键,与文献[2]值非常接近,且均在实验误差范围内。而对N—C 和C—H键,文献[3]收录的BDE 数据为1964年发表文献[15]中的数据,尤其对于N—C 键,原文献[15]采取与胺类化合物进行结构类比的方法得到N—C 键的BDE,而非实验测定值(未给出误差范围),所以其数值可信度更值得商榷。在甲基肼中,N—C 键BDE为(295.81±10.46)kJ/mol[2],偏二甲肼与甲基肼结构极其类似,推测其N—C 键的BDE 值应该非常接近,这与组合从头算方法所得结果(约260 kJ/mol)吻合。由于组合从头算方法具有很高的精度及其普遍成功性,本研究推荐以上组合从头算方法得到的数据作为偏二甲肼N—C 和C—H 键的BDE值。
组合从头算方法G3 和CBS-Q 与G3B3 和CBS-QB3 得到的数据差距很小,说明采用M P2 或B3LYP 方法进行几何构型优化对偏二甲肼BDE 计算影响不大,同时也可以说明得到的数据是可信的。
通过比较偏二甲肼中4 种类型化学键的BDE数据还可以发现,偏二甲肼分子内最弱的化学键为N—N 和N—C 键,推测偏二甲肼发生化学反应时容易发生这两个键的断裂,这与文献[15]结果一致。

表1 组合从头算方法的BDE 计算结果Table 1 Calculated BDEs by composite ab initio methods
2.2 密度泛函理论计算结果
分别利用限制性开壳层方法(RO)和非限制性(U)方法,采用B3LYP 方法,计算了偏二甲肼各个化学键的BDE,结果列于表2。通过与CBS-QB3 方法的计算结果进行比较发现,限制性开壳层方法得到的结果要明显优于非限制性方法。这是由于自由基是一种开壳层体系,非限制性计算方法存在自旋污染效应而引起的,因此推荐利用限制性开壳层方法进行相关BDE 的计算。计算结果显示,B3LYP 方法系统地低估了BDE,最大误差达到约40 kJ/mol。所以,B3LYP 方法不适合于计算偏二甲肼及与之类似结构化合物(如其他肼类物质,下同)的BDE,或必须经系统校正后才可以应用于键解离能的计算。
利用(RO)BM K 方法计算得到的偏二甲肼BDE 值见表2。比较发现,(RO)BM K 的计算结果非常接近组合从头算方法的结果,误差约为6 kJ/mol,且具有相对系统性,经系统校正后可以达到组合从头算方法的计算精度。因此,选择(RO)BMK 方法作为偏二甲肼及相关类似结构化合物键解离能的计算方法。
(RO)B3P86 方法对偏二甲肼N—H 和C—H键的解离能具有很好的计算结果,且稍优于BM K方法。对N—N 键解离能的计算结果与BM K 方法很接近,对N—C 键的计算结果则较差,与CBSQB3 的结果相差15 kJ/mol。因此(RO)B3P86 方法更适合计算偏二甲肼及类似结构化合物X—H 键的键解离能(X表示任意原子),与文献[15]结论一致。
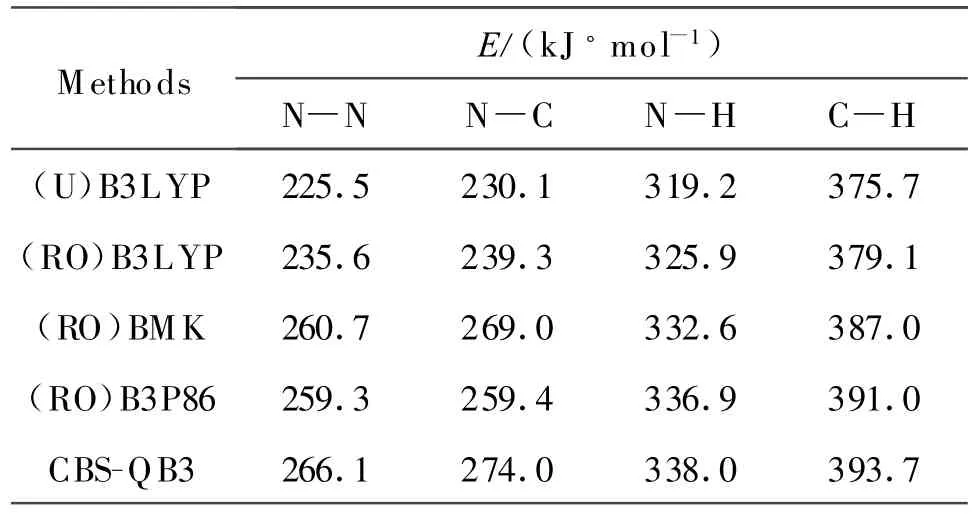
表2 用密度泛函理论方法和CBS-QB3 计算的BDE 值Table 2 Calculated BDEs by DFT and CBS-QB3
3 结 论
(1)文献[2]收录的N—C 和C—H 键解离能数据可能存在误差,推荐高精度的组合从头算方法得到的数据作为偏二甲肼N—C 和C—H 键BDE 的参考值。
(2)限制性开壳层方法所得结果要优于非限制性方法。B3LYP 方法的计算结果系统性地低估了BDE,(RO)BM K 方法的计算结果普遍较好,(RO)B3P86适合N —H 和C—H 键BDE 的计算。计算结果表明,偏二甲肼分子中N—N 和N—C 键的BDE 最低。
[1]李正莉,王煊军,张有智.偏二甲基肼亲核性的量子化学研究[J].火炸药学报,2007,30(5):49-52.
LI Zheng-li,WANG Xuan-jun,ZHANG You-zhi.The quantum chemical investigation on the nucleophilicity of UDMH[J].Chinese Journal of Explosives and Propellants,2007,30(5):49-52.
[2]罗渝然.化学键能数据手册[M].北京:科学出版社,2005.
[3]Curtiss L A,Raghavachari K,Redfern P C,et al.Gaussian-3(G3)theory for molecules containing first and second-row atoms[J].J Chem Phys,1998,109(18):7764-7776.
[4]Baboul A G,Curtiss L A,Redfern P C,et al.Gaussian-3 theory using density functional geomet ries and zero-point energies[J].J Chem Phys,1999,110(16):7650-7657.
[5]Ochterski J W,Petersson G A,Montgomery J A Jr.A complete basis set model chemistry.V.Extensions to six or more heavy atoms[J].J Chem Phys,1996,104(7):2598-2619.
[6]Montgomery J A Jr,Frisch M J,Ochterski J W,et al.A complete basis set model chemist ry.VII.Use of the minimum population localization method[J].J Chem Phys,2000,112(15):6532-6542.
[7]YU Yi-Yun,FU Yao,LIU Lei,et al.Theoretical study of remote substituent effects on X—H (X=CH2,NH,O)bond dissociation energies of azulene[J].Chinese J Chem,2007,25(7):1014-1022.
[8]S UN Hong-yan,Law Chung K.Thermochemical and kinetic analysis of the thermal decomposition of monomethylhydrazine:an elementary reaction mechanism[J].J Phys Chem A,2007,111(19):3748-3760.
[9]SONG Ke-sheng,LIU Lei,GUO Qing-xiang.Effects of geminal disubstitution on C -H and N -H bond dissociation energies[J].Tetrahedron,2004,60:9909-9923.
[10]Bohn M A,Klapotke T M.DFT and G2M P2 calculations of the N-N bond dissociation enthalpies and enthalpies of formation of hydrazine,monomethylhydrazine and symmetrical and unsymmetrical dimethylhydrazine[J].Z Naturforsch B,2004,59b:148-152.
[11]Becke,A D Density-functional exchange-energy approximation with correct asymptotic behavior[J].Phys Rev A,1988,38(6):3098-3100.
[12]Boese A D,Martin J M L J.Development of density functionals for thermochemical kinetics[J].J Chem Phys,2004,121(8):3405-3416.
[13]Perdew J P.Density-functional approximation for the correlation energy of the inhomogeneous electron gas[J].Phys Rev B,1986,33(12):8822-8824.
[14]Frisch M J,T rucks G W,Schlegel H B,et al.Gaussian 03.Revision E.01[M].Pittsburgh,PA:Gaussian Inc,2004.
[15]Eberstein I J.The gas phase decomposition of hydrazine propellants,AD-607334[R].New Jersey:Princeton University,1964.
