AMS测量32Si的ΔE-Q3D方法
2010-03-24李朝历陆丽燕李世琢
龚 杰 李朝历 王 伟 陆丽燕 李世琢 何 明 姜 山
1(中国原子能科学研究院 北京 102413)
2(广西大学物理科学与工程技术学院 南宁 530004)
32Si是Si的唯一长寿命放射性核素,半衰期为~140 a,在 100–1000 a 尺度的测年[1–3]和硅的生物地球化学循环研究[4,5]中具有极其重要的应用价值。自然界中的32Si是由初级或次级宇宙射线与空气中的Ar通过散裂反应产生,反应式为40Ar(n,4p5n)32Si、40Ar(p,2αp)32Si。32Si产生于上层大气圈,约 2/3产生于平流层,1/3产生于对流层,通过雨雪或干散落物的形式降落到地表的硅储存库中。自然界的32Si的产生率非常低,地球上平衡总量为~2 kg,其各种自然样品的天然同位素丰度一般低于10–14。
但受测量方法所限,目前尚无精确的、公认的32Si半衰期值[1,6],这极大地限制了32Si的应用。二十世纪80年代前,测量32Si采用放射性法[7]。32Si为纯β衰变,发射低能(0.22 MeV)β射线,直接进行测量较为困难,它衰变生成的子同位素32P(14.3 d),发射高能量(1.7 MeV)β射线。可测量32P的放射性得到32Si的放射性。但自然界中的32Si含量极低,大量的32Si难以获得,使这种方法受到限制。
上世纪 80年代发展起来的加速器质谱技术(AMS),测量灵敏极高度,所需样品量非常少,能对毫克级的样品在较短时间内完成分析,尤宜于同位素比很低的研究工作, 如32Si。国际上已用AMS进行过多次32Si测量,探测灵敏度好于10–15,也已进行自然样品的测量[8–12]。我国刘存富等[13]用放射性法测量了地下水中的32Si,尚未进行 AMS测量32Si的研究。
此外,生成32Si的反应道少、反应域高、反应截面小,大量制备高含量的32Si样品受实验条件、生产成本等的限制,这成为制约32Si广泛应用的另一重要因素。
人工生产32Si的方法主要有:
(1) Honda等[14]用730 MeV质子轰击Fe,通过散裂反应生产32Si。其反应截面非常小,能量如此之高的反应截面也仅0.46 mb,不适于生产32Si。
(2)36S(p,3p2n)32Si:30 MeV质子的反应截面约3 mb。但36S的天然丰度仅0.015%,36S富集成本较高,且对于AMS测量32Si,使用S靶无疑将引入同量异位素32S的干扰。
(3)37Cl(p,α2p)32Si:Elmore 等[15,16]曾用高能质子轰击37Cl生产32Si。质子能量大于35 MeV时反应截面约1 mb,低于35 MeV则截面急剧下降。
(4)31P(n,γ)32P(n,p)32Si:第一级反应31P(n,γ)32P的热中子截面为190 mb,第二级32P(n,p)32Si在中子能量大于1 MeV时截面120 mb。Hofmann[6]曾用此反应生产32Si。但第一级反应需热中子、第二级需1 MeV以上的快中子的特点,需有热快比好、通量高的中子源,宜用散裂中子谱辐照。伴随反应有31P(n,p)31Si(n,γ)32Si,Jantsch[17,18]在用 P 生产32Si的实验中发现,由该反应与31P(n,γ)32P(n,p)32Si反应产生的32Si分别为3.9%和2.8%。
(5)30Si(n,γ)31Si(n,γ)32Si,此反应只用热中子,两级反应的热中子反应截面分别为 107 mb和 180 mb[19],虽小于31P(n,γ)32P(n,p)32Si的反应截面,但可在热中子堆中辐照。
中国原子能科学研究院的强流质子回旋加速器可提供30 MeV质子束,此能量下可用富集36S进行辐照,但此法受实验成本制约并引入32S的强烈干扰,不适于AMS测量。此外还有30Si(t,p)32Si[22]、18O(16O,2p)32Si[19]等反应,但前者不易获得高通量的t束,后者则因18O的天然丰度仅0.2%而难以得到较高产额的32Si。
鉴此,我们选择用游泳池反应堆辐照31P和30Si生产32Si,选择了合适的反应生产32Si,建立了用于AMS测量的32Si样品制备化学流程,探索了AMS测量32Si的各种实验条件,最终建立了AMS测量32Si的DE-Q3D的方法,基于Q3D磁谱仪进行32S的排除,并利用多阳极气体电离室对其作进一步分辨。本文对上述进展作系统介绍。
1 实验方法
1.1 32Si的生产
在中国原子能科学研究院游泳池堆上利用31P(n,γ)32P(n,p)32Si和30Si(n,γ)31Si(n,γ)32Si反应生产32Si,辐照时间为250 h。表1为样品辐照的部分数据,图1为两种反应生成的32Si与受辐照元素的原子数之比随辐照时间的变化。现已完成样品辐照,并基本冷却完毕,准备进行化学处理,制作成适合AMS测量用的样品形式。

表1 样品辐照部分数据*Table 1 Part of the data of the sample radiation.
产额计算公式为:
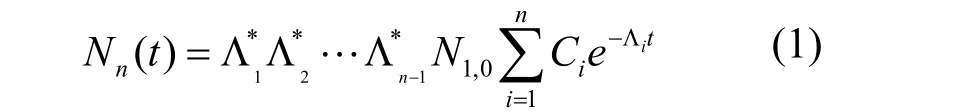
其中,Ci是消失系数,Ci= ∏j=1→n(Λj–Λi)–1, (i≠j);是转变系数,若第i+1个核仅由第i个核素衰变而来,若第i+1个核仅由第i个核素由核反应而来,Λi=Φiσi;若两者都有贡献,则
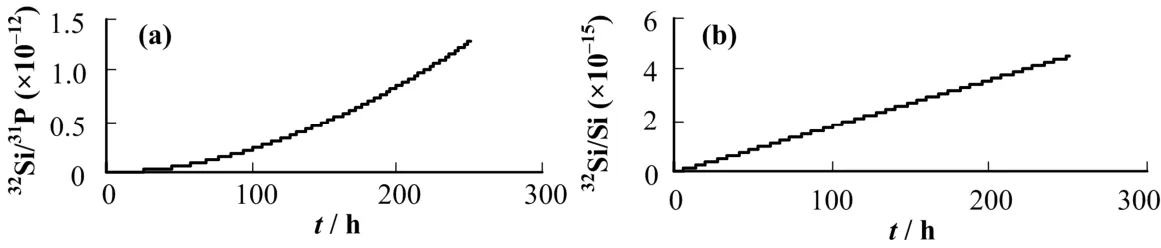
图1 Mg2P2O7(a)与SiO2(b)的辐照产额曲线Fig.1 The irradiation yield curve of Mg2P2O7 (a) and SiO2(b).
1.2 离子源引束
为确定合适的样品化学形式、导电介质种类及掺杂比例、离子源引出形式等,我们进行了一些引束实验。实验比较了单质 Si、Mg2Si、SiO2在相同条件下引出的28Si-束流大小,结果显示单质Si>SiO2>Mg2Si。但是由于单质Si的制备非常复杂,故选择SiO2作为离子源样品形式。
当SiO2以不同比例分别掺杂Al、Ag、Fe、Nb、Cu粉作为导电介质放入负离子源引28Si-束流时,掺Nb和Fe粉引出了最大的束流,并且在质量比为1:5 (SiO2:Fe/Nb=1:5)时得到的束流最大(图2)。实验还测量了SiO2:PdF2=1:5时引出的SiFx–束流情况(图3a)和掺不同导电介质时SiOx–的引出情况(图3b)。
实验结果表明,引出的氟化物及氧化物束流都比较小,不适合用作 AMS测量。经比较,选定样品化学形式为SiO2,按质量比1:5掺Fe粉为导电介质,离子源引出形式为Si-。
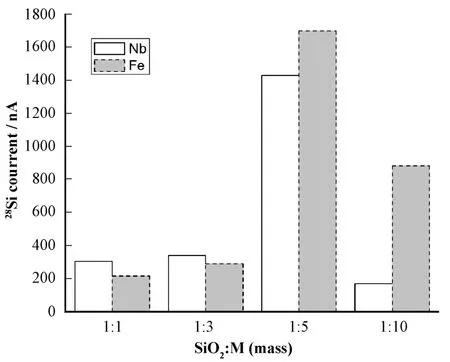
图2 导电介质为Fe、Nb时,28Si-束流比较Fig.2 28Si- beam current using Fe and Nb as conducting medium.

图3 SiO2 : PdF2=1:5时引出的(a) SiFn- 与(b) SiOn-束流Fig.3 Beam current of (a) SiFn- and (b) SiOn- at SiO2 : PdF2=1:5.
1.3 样品制备
经过多次的探索和摸索条件,建立了以下的样品化学处理流程(图4)。

图4 Mg2P2O7中的32Si的化学分离流程装置图Fig.4 The apparatus for chemical separation of 32Si in Mg2P2O7.
1.3.1 辐照样品分离流程
将Mg2P2O7溶于HF酸,加入Si载体(Na2SiO3),再加入KF溶液,得到K2SiF6沉淀。
K2SiF6在沸水中会水解:

向水解溶液滴加HNO3使H2SiO3沉淀,在150℃以上加热H2SiO3使之分解,得到SiO2。
1.3.2 高含量样品稀释流程及排硫流程
1.3.2.1 碱溶法
实验中所用32Si样品由澳大利亚国立大学提供,其为32Si/Si=4×10–10的SiO2样品。该样品经图5 碱溶法流程稀释为32Si/Si=4×10–11、4×10–12、4×10–13、4×10–14、4×10–15的系列样品。
碱溶法实验中,SiO2全部溶解后才能进行下一步,沉淀过程中则需调节pH值,pH过低会将刚形成的SiO2分子重新溶解。

图5 SiO2标样稀释碱溶法流程Fig.5 The alkali solubilization procedure of SiO2 standard sample dilution.
1.3.2.2 酸溶法
将高32Si含量SiO2样品及稀释载体用HF酸溶解,用氨水沉淀,得Si(OH)4,放入烘箱在200℃下分解,得SiO2,用稀硝酸、去离子水和无水乙醇反复清洗,放入马弗炉,在1100℃下灼烧,以去除样品中的S。
相比于碱溶法,酸溶法实验周期短、操作简单、回收率高,但所得SiO2颗粒较粗,需要经研磨才能用于AMS测量;所得SiO2样品中的S含量也高于碱溶法。该方法是新建立的稀释方法,正对其进行改进,以期替代碱溶法。
1.4 AMS测量
实验在中国原子能科学研究院的HI-13型串列加速器Q3D大型磁谱仪(图6)上进行。
AMS测量32Si受其同量异位素32S(天然同位素丰度 95%)的强烈干扰。此DE-Q3D方法基于 Q3D磁谱仪进行32S排除。样品置于负离子源中,负离子束流从离子源引出后经磁、电偏转后只留下质量32、电荷–1的离子,送入加速器加速。在加速器中部经C膜剥离,基于库仑爆炸效应排除分子离子的干扰,并剥离出高电荷态继续加速。加速器后的高能离子经高能分析系统的偏转后只留下质量与电荷数都相同的同量异位素32Si和32S的束流,传输至Q3D磁谱仪。Q3D磁谱仪前有一层剥离膜。Si与S的质子数不同,它们穿过该剥离膜后具能量损失有异,它们的剩余能量差异使它们在磁谱仪中的偏转路径不同,从而排除32S。然而,由于能量离散,部分32S仍会落在32Si区域,为排除这一干扰,我们在 Q3D磁谱仪的焦面处利用多阳极气体电离室对其作进一步分辨。
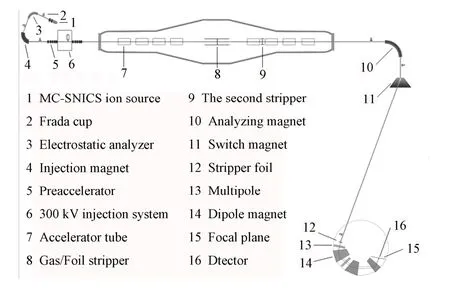
图6 串列加速器-Q3D设备示意图Fig.6 Schematics of the tandem accelerator-Q3D facilities.
在 Q3D磁谱仪前的靶室内使用的是 3 μm的Si3N4膜。这种膜密度大,束流穿过后能散小,使32Si与32S具有更大的能损差,更好地将它们分开,又不至于造成很大能散(表2)。

表2 84 MeV 32Si和32S离子穿过不同厚度Si3N4膜后剩余能量Table 2 Residual energy of 84 MeV 32Si and 32S ions penetrating Si3N4 foils of different thicknesses.
多阳极气体电离室探测器是目前最为成熟的探测器之一,可获得离子在不同射程时的能量损失及其剩余能量,利用其形成的单维谱和双维谱对32Si和32S进行鉴别。
我们用与32Si质量最接近的稳定同位素30Si模拟32Si的光路。加速器端电压10.48 MV,C膜剥离,过C膜后选择7+电荷态(剥离几率34.68%),则32M在高能端的能量为 83.95 MeV。过 Si3N4膜后选择11+电荷态(剥离几率30.8%)。电离室位于Q3D焦面中心位置,内充C4H10气体,由流气系统将气压稳定在42 mbar。在焦面处还装有可上下左右移动的金硅面垒型半导体探测器(Au-Si SBD,Φ20 mm,上升时间14 ns,北京核仪器厂生产),测量不同位置的计数及刻度能量。

图7 32Si和32S在电离室中的比电离曲线Fig.7 Specific ionization curve of 32Si and 32S in chamber.
实验中的32S干扰强,除在样品制备过程中尽可能降低样品S含量外,还要在测量参数选择上作此努力,如Q3D剥离电荷态选择中,未用32Si剥离几率最高(44.771%)的32Si12+,而用32Si11+,因为与32S的剥离几率比较,后者可将32S多压低一个量级。
我们使用四阳极电离室,四块阳极板长度分别为100、80、60、126 mm。为提高电离室的灵敏度,选取适当电离室气压使32S的绝大部分能量损失在第三阳极板上。32Si与32S的比电离曲线(图7)则在第二或第三阳极板交叉。表3为32Si和32S在电离室中的能损比较。
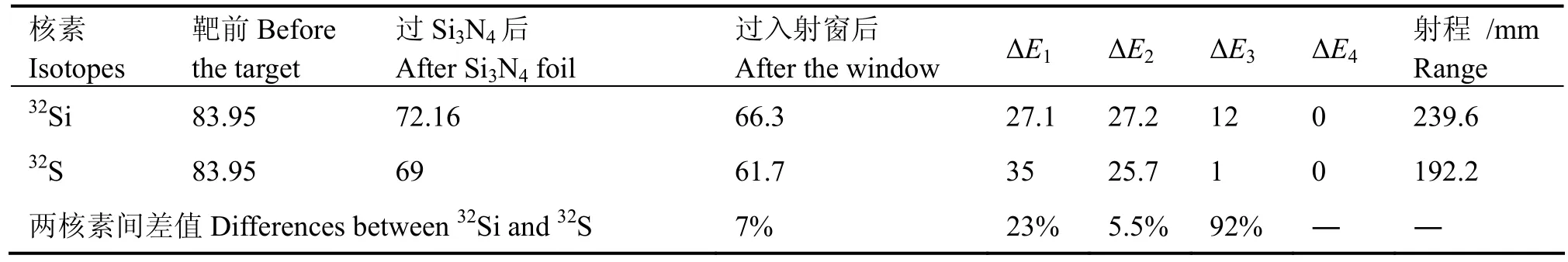
表3 32Si和32S在电离室中的能损比较Table 3 The energy loss comparison between 32Si and 32S in chamber.
由于SRIM软件模拟存在一定误差,同时Q3D具有大色散的特点,使32Si在焦面位置的模拟与真实情况存在偏差。而32Si含量非常低,32S计数却非常强,32Si在Q3D焦面的计数很易被32S的散射本底掩盖,用半导体探测器将无法确定其峰位。鉴此,我们使用了如下精确确定32Si在焦面位置的方法:
(1) 在11 MV端电压下用30Si通光路,在Q3D不挡 Si3N4膜的情况下将其传至焦面,这时其能量是精确已知的。利用Au-Si SBD谱仪寻找其峰位。
(2) 由已测得的36S数据,99.1 MeV 峰位为655.7道,则 Au-Si SBD谱仪的能量刻度为 E=0.154–1.878。
(3) 将30Si穿过 3 μm Si3N4膜后寻找其11+峰位,调节Q3D磁场将其偏转至中心位置(即电离室入射窗位置)。测得到其在512道,即能量为76.97 MeV,则与其具有相同磁刚度的32Si能量应为72.16 MeV。用SRIM计算在过3 μm Si3N4膜后84 MeV的32Si应比88 MeV30Si多损失0.55 MeV能量,所测过3 μm Si3N4膜后30Si的能损为11.24 MeV,故在靶前32Si能量应为 83.95 MeV, 对应端电压为10.48 MV。
这样,端电压设为10.48 MV,32Si束流可准确地进入电离室。目前,我们已用上述方法对32Si样品进行了三次 AMS测量,成功建立了加速器质谱测量32Si的DE-Q3D方法。
2 结果和讨论
由于没有标准样品,从澳大利亚国立大学获得的样品虽有标称值,但无法通过其它方法进行验证,因此本实验采用绝对测量。
实验中加速器端电压选取10.48 MV,32Si能量83.95 MV,加速器C膜剥离,电荷态选取7+,Q3D剥离膜选择 3 μm Si3N4膜,整个实验传输效率为3.9% (L.E, 靶前),测量效率1.2%。图8为对Q3D焦平面用Au-Si SBD谱仪扫描得到的计数曲线。

图8 Q3D焦面计数Fig.8 The counts on the Q3D focal plane.
图8中,电离室的位置固定,束流位置可由调节Q3D的磁场左右偏转。Au-Si SBD安装在电离室入射窗前,可左右移动。32S和32Si经过Q3D的靶室后由于剩余能量差异而落在不同的焦面位置。32Si的位置经前述测量方法确定,32S的主峰位已被偏离出电离室探测器,32Si和少量的32S将进入电离室探测器。
实验中低能端30Si束流为22.9 nA(同位素丰度3.10%),测量时间50 min,电离室采用流气系统,内充异丁烷,自动稳压在42 mbar,使32S被全部阻止在第三阳极板前,32Si能达到第四阳极板。图 9为电离室得到的双维谱,E1-Er和 Et-Er谱可很好地看到32Si计数。
谱中的计数为15个。计数所处的Q3D焦面的位置和双维谱的能量区域,都与理论计算得到的32Si的位置和能量区域一致。实验中,调节Q3D磁谱仪的主二极磁铁的磁场值,从而改变束流落在焦面的位置,测量束流位置左右各5 cm内的焦面计数,均未在32Si的能谱区域内得到计数。由此可判定谱中计数就是32Si。从而计算得到样品中的32Si/Si=9.03´10–14,与样品的标称值 1×10–13相差9.7%,两者基本一致。
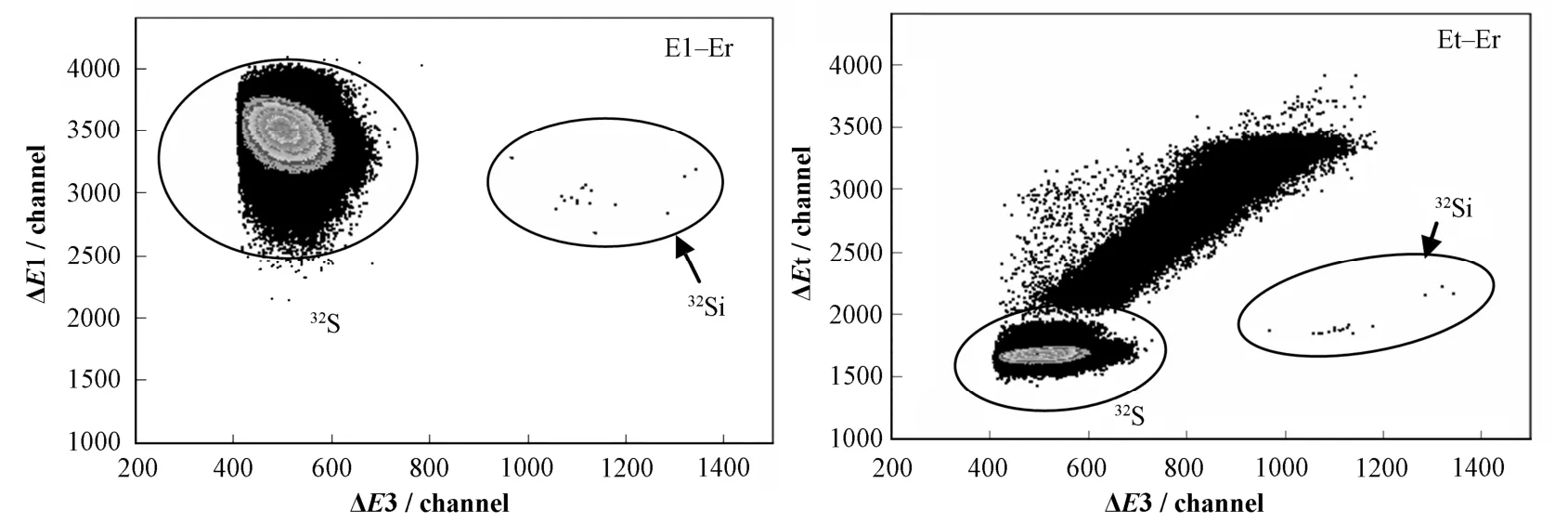
图9 电离室得到的双维谱Fig.9 The two dimensional spectra got by the ionization chamber.
由图 9,尽管在进入电离室以前已通过各种方法排除了样品中的绝大部分32S,但与32Si相比,S的计数仍然非常强。Au-Si SBD谱图中,32Si的计数被32S的“尾巴”覆盖而无法鉴别。32Si的AMS测量研究中,32S的干扰是一个无法避免的问题。经计算,化学流程将S压低104,使用Q3D加能量损失膜对32S的压低因子为105,气体电离室对84 MeV的32Si和32S的压低因子预计105,总的探测限达到10–14。总体说来,32S的干扰被控制在可接受的水平。但若束流进一步提高,有可能因为32S的计数太强而使得电离室探测器无法正常工作。
在对一个空白样品的测量中,我们经过20 min的测量在32Si的能谱区域没有得到计数,测量灵敏度好于 1.5×10–14。
3 结论
本工作侧重于实验方法的建立。成功测到了32Si的计数,样品测量值与标称值误差好于10%,测量灵敏度好于1.5×10–14,成功建立了加速器质谱高灵敏测量32Si的DE-Q3D方法。
但是实验中32S的干扰仍较严重。我们将继续探索排S化学、物理方法,针对绝对测量中的效率问题展开仔细研究,寻找误差来源,不断改进和优化实验方案,研制自己的标准样品并对我们的辐照样品和一些自然样品进行测量,以提高实验的灵敏度和准确性。
1 Nijampurkar V N, Amin B S, Kharkar D P.Nature, 1966,210:478–490
2 Bhandari N, Nijampurkar V N, Vohra C P.Pro Symp on Variations in the Global Water Budget, 1981.207–220
3 Nijampurkar V N, Rao D K.Snow and Glacier Hydrology,1993, 218: 355–369
4 Craig H, Somayajulu B L K, Turekian K K.Earth Planet Sci Lett, 2000, 175: 297–308
5 Lal D, Nijampurkar V N, Somayajulu.Limnology and Oceanpgerapity, 1976, 21(2): 285–293
6 Chen Y, Kashy E, Bazin D, et al.Phys Rev C, 1993,47(4):1462–1465
7 Alburger D E, Harbottle G, Norton E F, et al.Earth Planet Sci Lett, 1986, 78: 168–176
8 Uwe Morgenstern, Fifield L Keith, Zondervan Albert.Nucl Instr Meth B, 2000, 172: 605–609
9 Kutschera W, Henning W, Paul M, et al.Phys Rev Lett,1980,45: 592–596
10 Thomsen M S, Heinemeier J, Hornshoj P, et al.Nucl Phys,1991, A534: 327–338
11 Zoppi U, Kubik P W, Suter M, et al.Nucl Instr Meth B,1994, 92: 142–145
12 Treacy Jr D J, Knies D L, Grabowski K S, et al.Nucl Instr Meth B, 2000, 172: 321–327
13 万君伟, 刘存富, 姚念英.同位素水文理论与实践.武汉: 中国地质大学出版社, 2003.85–97 WAN Junwei, LIU Cunfu, YAO Nianying.Theory and practice of isotope hydrology.Wuhan: China University of Geosciences Press, 2003.85–97
14 Masatake Honda, Devendra Lal.Nucl Phys, 1964, 51:363–368
15 Elomre D, Anantaraman N, Fulbright H W, et al.Phys Rev Lett, 1980,45: 589–592
16 Hofmann H J, Bonani G, Suter M, et al.Nucl Instr and Meth B, 1990, 52: 544–551
17 Jantsch, Kernenergie, 10.Jahrgang, Heft 1967, 3: 89
18 Jantsch, Kernenergie, 9.Jahrgang, Heft 1966,4: 127
19 Thomsen M S, Heinemeier J, Hornshoj P, et al.Nucl Instr and Meth B, 1988, 31:425–432
