咪唑啉衍生物缓蚀性能的密度泛函理论和分子动力学模拟
2010-01-30胡松青胡建春郭文跃
胡松青,胡建春,张 军,石 鑫,郭文跃
(中国石油大学物理科学与技术学院,山东东营257061)
腐蚀是困扰我国油气工业发展的一个极为突出的问题。在大量的实践中,人们根据被腐蚀材质的特点及腐蚀介质的不同,发现了许多实用而有效的防腐方法。采用缓蚀剂作为一种经济有效的防护技术广泛地应用于石油、化工、能源、交通、建筑等工业部门,并成为不可替代的重要防护措施。缓蚀剂的合成和筛选目前仍以大量实验为基础,直接根据腐蚀环境设计和合成的例子还不多见。因此,研究缓蚀剂的缓蚀作用机理,为缓蚀剂的设计提供理论指导具有重要的意义。量子化学方法在研究有机分子结构、性能和反应活性等方面获得了很大的成功。自从 Vosta等[1]于 1971年用 HMO(Hiickel molecular orbital method)分子轨道近似方法研究有机缓蚀剂以来,量子化学计算方法已成为研究吸附型缓蚀剂分子结构与其缓蚀性能关系的有效手段[2-5]。近年来,随着计算机技术的发展,关于缓蚀剂缓蚀机理的研究开始从静态向动态、从小体系向介观尺度过渡。一些研究人员[6-8]开始了缓蚀剂缓蚀机理的分子动力学模拟研究,分析温度、溶剂性质等因素对缓蚀剂在金属表面吸附行为的影响,为研发和评价新型高效缓蚀剂提供了直接依据。
量子化学计算和分子动力学模拟在缓蚀剂的理论研究中体现出巨大的优势,并逐渐成为该领域理论研究的重要工具[9]。但总体来看,关于缓蚀剂缓蚀机理的理论研究还比较薄弱,没有形成统一的理论体系。目前,在缓蚀剂缓蚀机理的研究中,量子化学计算和分子模拟2种方法相互独立,将两者相结合进行该研究的仍不多见。鉴于此,笔者以自制的4种咪唑啉类有机缓蚀剂为主要研究对象,采用量子化学计算和分子模拟相结合的方法,从不同层次系统地研究缓蚀剂分子结构特征及缓蚀剂与金属表面的作用机制,深入探讨有机缓蚀剂分子结构和其缓蚀效率的关系,分析总结缓蚀剂在金属表面的吸附、成膜规律,为设计开发新型、高效、低毒环保的有机缓蚀剂提供理论指导。
1 实验和理论方法
1.1 实验用缓蚀剂
采用自制的4种咪唑啉类有机缓蚀剂,其名称和分子结构示于表1。

1.2 失重法
选取尺寸为50 mm×10 mm×1 mm的Q235A钢试样若干,将其表面用 180#、380#、800#、1000#、1200#、2000#水砂纸逐级打磨,然后用清水冲洗试样表面,再用丙酮、无水乙醇依次对试样进行脱脂并清洗,在Cortest公司生产的34.4 MPa高温高压釜中进行失重法实验。实验所用腐蚀介质为含饱和CO2的3%(质量分数)NaCl溶液,缓蚀剂(A、B、C、D)用量为100 mg/L。基本实验温度为298 K,腐蚀时间72 h。实验结束后用电子天平称其质量,按公式(1)计算缓蚀效率 IE。式(1)中,W0和 Wt分别为未加和加入缓蚀剂失重实验后试样质量的改变量,g。为了尽量减小误差,采用平行实验求得 IE平均值。
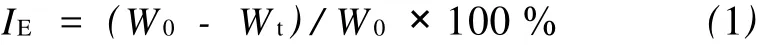
1.3 量子化学计算
由 Gauss View软件构建咪唑啉衍生物 A、B、C、D的分子初始构型,然后在 Gaussian 03软件包中,采用密度泛函理论(DFT)中的B3L YP方法,在6-31G*基组水平上进行几何构型优化,并进行频率分析,确保所得的结构均为势能面上的极小点(无虚频);在同一基组水平上计算分子的前线轨道分布、偶极距和Fukui指数,用于分析缓蚀剂的活性和选择性。
1.4 分子动力学模拟
通过量子化学方法可以构建缓蚀剂缓蚀性能与其分子结构的关系,对缓蚀机理进行研究。但是,缓蚀剂的缓蚀性能除自身反应性之外,还与具体的腐蚀环境有关。通过计算机模拟“缓蚀剂分子-腐蚀界面”体系达到平衡时的构型,获取界面上的吸附能数据,对于评价缓蚀剂性能的好坏具有重要的意义。缓蚀剂分子在 Fe表面的吸附能可由式(2)给出[10]。
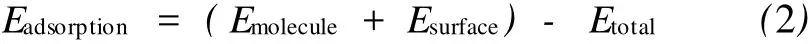
式(2)中,Eadsorption为吸附能;Emolecule是自由分子的能量;Esurface是未吸附分子时金属表面的能量;Etotal是包含1个分子和金属表面体系的总能量。
采用分子动力学 (MD)方法模拟缓蚀剂与 Fe表面的相互作用。选取 Fe晶体的(001)晶面为吸附表面,构建1个包含11层共计1734个Fe原子的表面体系,其大小为3.44 nm×3.44 nm×6.43 nm。然后用Charge group方法定义几何构型优化好的缓蚀剂分子,并导入到 Fe表面上。
计算中“冻结”表面体系中所有原子,而吸附分子保持与金属表面自由相互作用。先采用 Material Studios 4.0中的 Compass力场对“吸附分子-Fe”体系进行优化,再采用 Discover模块进行动力学模拟。模拟中采用NVT恒定的正则系统,模拟温度298 K,采用 Charge group方法计算范德瓦尔斯和库仑相互作用,截断半径1.5 nm,时间步长1 fs,总模拟时间5×10-7s,每隔2×10-9s采集1次构型。
2 结果与讨论
2.1 失重法实验结果
采用失重法测得的4种缓蚀剂在 CO2饱和的3%NaCl溶液中的缓蚀效率列于表2。从表2可以看出,这4种咪唑啉衍生物缓蚀剂都具有很好的抗CO2腐蚀性能,其中B的缓蚀效率最高,在其添加浓度为100 mg/L时缓蚀效率可达97.3%。
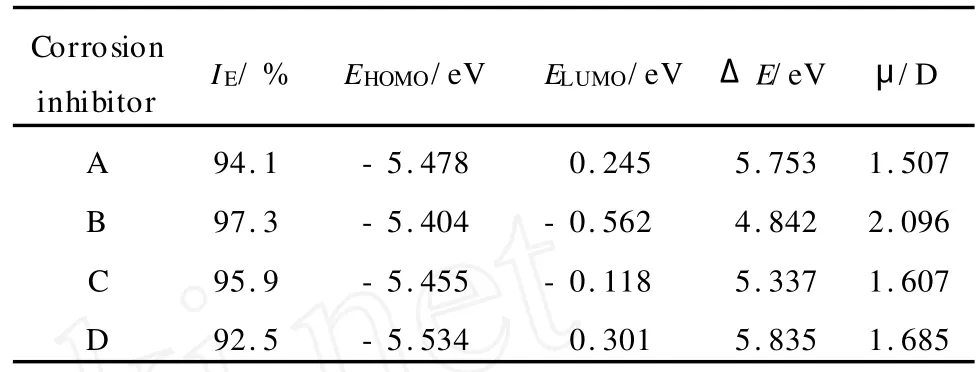
表2 4种缓蚀剂的缓蚀效率(IE)和量子化学参数Table 2 Inhibition efficiencies(IE)and quantum chemistry parameters of four corrosion inhibitors
2.2 量子化学计算结果
2.2.1 缓蚀剂分子优化的最终构型
4个咪唑啉类缓蚀剂分子在B3L YP/6-31G*理论水平上优化的最终构型如图1所示(原子已标号),其频率计算没有负值,表明各分子均达到基态。利用 Gauss view软件可以得出优化后各缓蚀剂分子咪唑环中原子之间的键长和键角信息,它们与已报道[11-12]的咪唑啉衍生物结构参数的实验数据基本相符,说明本研究中对缓蚀剂分子结构的优化所采用的方法可靠。

图1 A、B、C、D 4种缓蚀剂分子在 B3LYP/6-31G*理论水平优化的几何构型Fig.1 Optimized structures of A,B,Cand D corrosion inhibitor molecules at B3LYP/6-31G*level
2.2.2 缓蚀剂量子化学参数与缓蚀性能的关系
在量子化学的密度泛函方法对分子几何全优化的基础上,计算了缓蚀剂分子的偶极矩、最高占据轨道能量 EHOMO、最低空轨道能量 ELUMO及分离能ΔE,结果也列于表2。
多数有机缓蚀剂在腐蚀介质中对腐蚀的抑制是由于缓蚀剂吸附于金属表面的结果[13]。一种缓蚀剂的缓蚀效率主要决定于其分子在金属表面吸附作用的强弱,吸附作用分为物理吸附和化学吸附两部分。为探讨所涉及的缓蚀剂分子与金属表面的作用机制,对表2所列缓蚀剂量子化学参数与缓蚀效率的关系进行了分析。将 EHOMO、ELUMO、ΔE和μ与缓蚀效率 IE进行一元线性回归拟合,得:

其中 r为相关系数。
在物理吸附中,当缓蚀剂分子的极性增大时,其与金属表面的原子应有较强的激化作用,缓蚀剂的缓蚀效率应有所增加。但式(6)所示缓蚀剂的缓蚀效率与分子偶极矩的相关系数 r=0.412,相关性并不好,表明缓蚀剂分子的偶极矩对其缓蚀性能影响不大,故这4种缓蚀剂分子在金属表面的吸附主要为化学吸附。
量子化学的前线轨道理论[14]认为,分子的最高占据轨道的能量 EHOMO是分子给电子能力的量度, EHOMO越高,该轨道中的电子越不稳定,则该分子越易提供电子参与亲核反应。分子的最低空轨道的能量 ELUMO与分子的电子亲和能有关,其值越低,该分子则有较强的接受电子的能力。分离能ΔE (ΔE=ELUMO-EHOMO)也是分子稳定性的重要指标。从式(3)可见,缓蚀剂的缓蚀效率与 EHOMO的相关性很好,达到0.967,表明缓蚀剂分子的缓蚀效率随其最高占据轨道能量的升高而升高,这是由于随缓蚀剂分子最高占据轨道能量升高,其提供电子与金属的空 d轨道成键能力增强,从而使得其缓蚀性能随分子最高占据轨道能量升高而增强。从式(4)可见,缓蚀剂的缓蚀性能与 ELUMO的相关性也较好,表明缓蚀剂在金属表面与金属原子作用时,缓蚀剂分子既可以向金属原子的空轨道提供电子,又可以从金属接受电子到缓蚀剂分子最低空轨道上,亦即缓蚀剂分子可同时作为亲核试剂和亲电试剂与金属作用。
图2为缓蚀剂A、B、C、D分子的最高占有轨道(HOMO)和最低空轨道(LUMO)的0.02a.u.等值面图。从图2可看出,A、B、C、D分子的前线轨道都分布在咪唑环和极性官能团上,说明缓蚀剂与金属表面发生吸附时,主要是咪唑环和极性官能团起作用;B和C分子的HOMO和LUMO有相同的分布中心,都集中在咪唑环上,这种分布可使吸附发生时咪唑环优先朝向金属表面,既有利于金属表面空 d轨道接受缓蚀剂分子提供的电子形成配位键,又有利于缓蚀剂分子利用其反键轨道接受来自金属表面的电子形成反馈键,使B、C分子在金属表面形成稳定的吸附;另外,咪唑环和极性官能团的优先吸附使具有疏水特性的烷基支链R在金属表面形成保护膜,从而阻碍腐蚀介质向金属表面扩散,使缓蚀性能进一步提高。
2.2.3 缓蚀剂分子的反应活性部位
为进一步研究A、B、C、D缓蚀剂分子的反应活性位点,使用密度泛函的B3L YP/6-31G*方法,在对分子几何结构全优化的基础上,进行了Mulliken布局分析计算。表3列出了由Mulliken分布分析结果计算的缓蚀剂分子的 Fukui指数。Fukui指数是研究有机化合物的亲电或亲核反应性以及确定分子的活性部位的有效方法[15]。表3中, fi(r)+和 fi(r)-分别为亲核 Fukui指数和亲电Fukui指数,表示分子中原子 i得到/失去1个电子时,原子上的电荷变化,其值越大表明该原子越易得到/失去电子。
通过对 Fukui指数的分析可以发现,A、B、C、D缓蚀剂分子的咪唑环上的8N、9N上 fi(r)-最大,表明8N、9N是亲电子进攻中心,可向金属提供电子形成配位键,而4种分子的亲核反应中心(fi(r)+最大)则比较分散,B、C缓蚀剂分子的亲核反应中心分布在咪唑环上,而 A、D缓蚀剂分子的亲核反应中心分布在极性官能团上。Fukui指数的分析结果与上面的前线分子轨道分析结果一致。
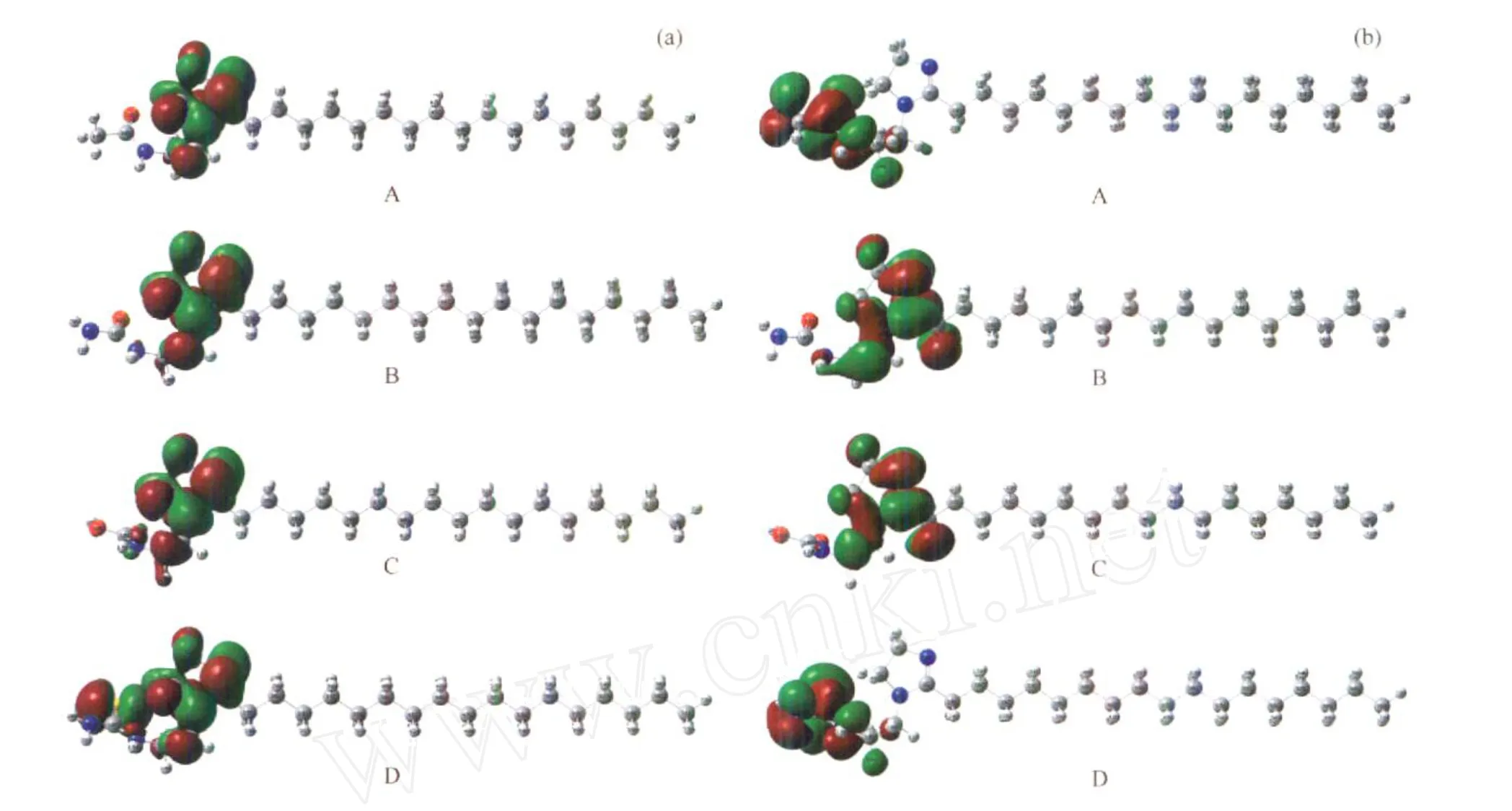
图2 缓蚀剂A、B、C、D分子的最高占有轨道(HOMO)和最低空轨道(LUMO)的0.02a.u.等值面图Fig.2 HOMO and LUMO isosurfaces with the value of 0.02a.u.for molecules of A,B,Cand D corrosion inhibitors (a)HOMO;(b)LUMO
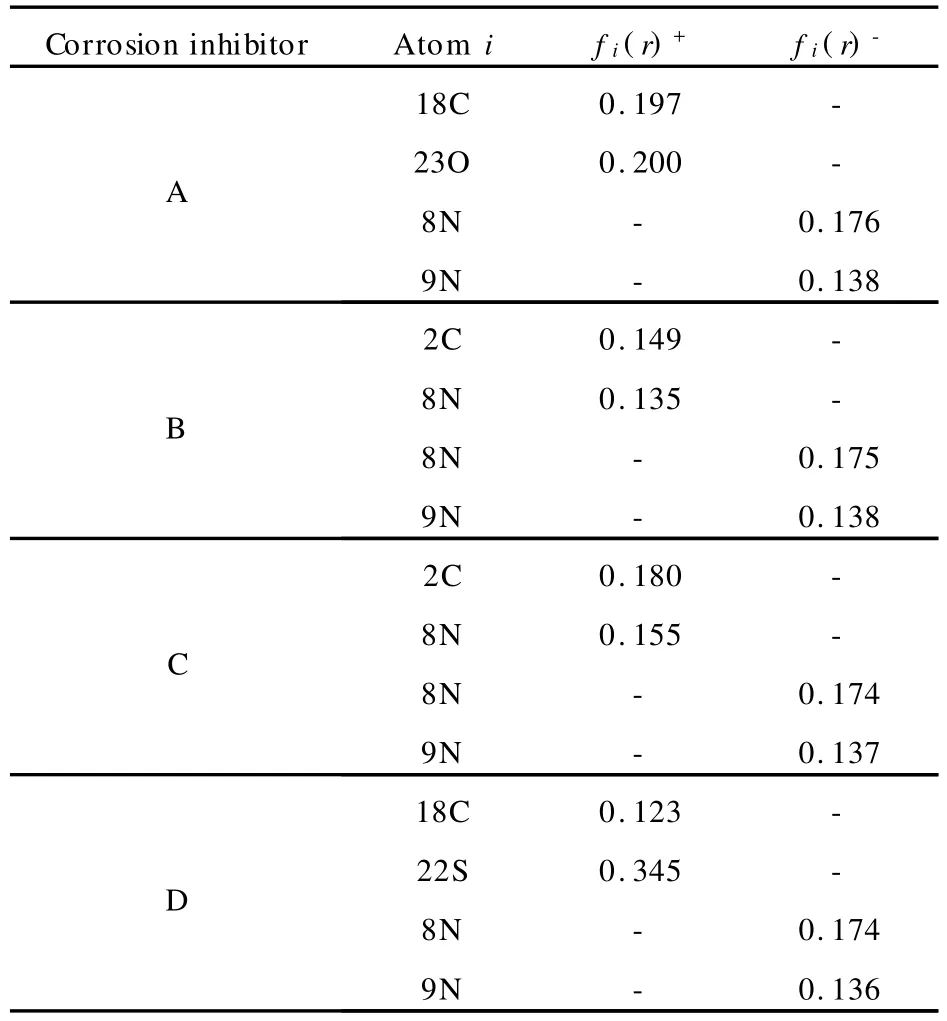
表3 4种缓蚀剂分子的Fukui指数Table 3 Fukui index values for the four corrosion inhibitor molecules
2.3 分子动力学模拟结果
2.3.1 吸附体系的平衡
对于平衡态的分子力学模拟,体系能否达到平衡至关重要。体系的平衡必须是温度和能量同时达到平衡。图3为A分子在 Fe(001)晶面上的温度和能量随时间的变化曲线。由图3(a)可见,体系温度基本在(298±10)K波动,标准偏差在3%左右,可以认为体系已经达到温度平衡。从平衡过程中所存的250帧轨迹看,体系能量波动亦渐趋平衡。从图3(b)可以看出,3×10-7s后体系能量波动平缓,各帧能量偏差仅为0.005%左右。由此可见,A分子在Fe(001)晶面上的吸附体系,经分子动力学模拟确已达到平衡,完全可以满足平衡态分子动力学模拟计算的要求,因此后面的计算、分析结果是完全可靠的。其它3种咪唑啉缓蚀剂分子与 Fe(001)晶面的相互作用也可得到相同的结论。
2.3.2 缓蚀剂分子在 Fe(001)晶面上的吸附能
通过分子动力学模拟,可以得到A、B、C、D及 H2O分子在 Fe表面的平衡吸附构型,如图4所示。从模拟过程中发现,无论放在金属表面的缓蚀剂分子的初始构型如何(缓蚀剂分子垂直、倾斜或平行于Fe表面),其咪唑环及头部的极性官能团由于较强的电荷转移作用,总是优先吸附于 Fe表面;而分子中烷基链则随咪唑环的吸附被逐渐“牵引”俯卧于Fe表面,这与上述分析HOMO和LUMO分布所得出的结论相吻合。体系达到平衡后,缓蚀剂分子在其平衡位置附近作微小振动,各分子的咪唑环均倾向于与Fe表面平行吸附。
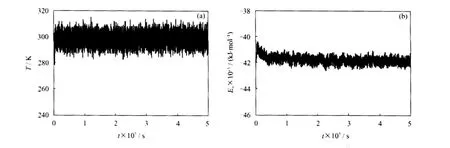
图3 缓蚀剂A分子在Fe(001)上吸附的温度平衡曲线和能量波动曲线Fig.3 Temperature equilibrium curve and energy fluctuant curve of A molecule on Fe(001)surface (a)Temperature equilibrium curve;(b)Energy fluctuant curve

图4 A、B、C、D及 H2O分子在Fe表面的平衡吸附构型Fig.4 Adsorption configurations on Fe surface of A,B,C,D and H2O molecules
缓蚀剂分子与金属表面的结合强度是衡量其缓蚀性能的一个重要指标[9],吸附能则是结合强度的最直接体现。经分子动力学模拟,得到A、B、C、D及 H2O分子与 Fe表面体系的相互作用能——单分子吸附能,其值分别为1336.590、1484.942、1551.415、1254.670、97.952 kJ/mol。可见4种缓蚀剂分子在Fe表面的吸附能都远大于 H2O分子的吸附能,说明A、B、C、D与 H2O分子相比能更稳定地吸附在金属表面,驱替金属表面的水分子,从而起到减缓腐蚀的作用。吸附能越大,缓蚀剂分子与Fe表面的相互作用越强,缓蚀性能也越好。因此,从单分子吸附能上看,4种缓蚀剂分子的缓蚀效率从大到小依次为B、C、A和D。这个结果进一步验证了上述量子化学计算的分析结果。
3 结 论
利用挂片失重法测得了A、B、C、D 4种咪唑啉衍生物缓蚀剂在 CO2饱和的3%质量分数 NaCl溶液中的缓蚀效率,并采用量子化学计算和分子动力学模拟相结合的方法对它们的缓蚀性能进行了理论评价。量子化学参数分析结果表明,缓蚀剂分子与金属界面作用时,主要是咪唑啉环和极性基团起作用,分子的反应活性中心主要集中在咪唑啉环和极性基团的 N、O、S原子处。单分子吸附的分子动力学模拟结果显示,4种缓蚀剂分子在 Fe表面的吸附能从大到小依次为B、C、A和D,吸附能越大,说明缓蚀剂分子与 Fe表面的相互作用越强,缓蚀性能也越好,与实验结果一致。
[1]VOSTA J,ELIASEK J.A quantum chemical study of the corrosion inhibition of iron by means of aniline derivatives in hydrochloric acid[J].Corrosion,1976,32 (5):183-185.
[2]YANG X C,ZHAO H,LI M D,et al.Quantum chemical study of the inhibition properties of pyridine and its derivatives at an aluminum surface[J].Corrosion Science,2000,42(4):645-653.
[3]FANG J,LI J.Quantum chemistry study on the relationship between molecular structure and corrosion inhibition efficiency of amides[J].Journal of Molecular Structure(Theochem),2002,593(3):179-185.
[4]KHALIL N.Quantum chemical approach of corrosion inhibition[J].Electrochimica Acta,2003,48(18): 2635-2640.
[5]RAAFAT M I,MOHAMEDK A,FATEN M A. Quantum chemical studies on the inhibition of corrosion of copper surface by substituted uracils[J].Applied Surface Science,2008,155(7):1-9.
[6]RAMACHANDRAN S,JOVANCICEVIC V.Molecular modeling of the inhibition of mild steel carbon dioxide corrosion by imidazolines[J].Corrosion,1999,55(3): 259-267.
[7]DU H,MILL ER J D.A molecular dynamics simulation study of water structure and adsorption states at talc surfaces[J].Int J Miner Process,2007,84(1-4):172-184.
[8]张曙光,陈瑜,王风云.苯并三氮唑及其羧酸酯衍生物对铜缓蚀机理的分子动力学模拟研究[J].化学学报, 2007,65(20):2235-2242.(ZHANG Shuguang,CHEN Yu,WANG Fengyun.Molecular dynamics simulation of the corrosion inhibition mechanism of copper by benzotriazole and its carboxylate derivatives[J].Acta Chimica Sinica,2007,65(20):2235-2242.)
[9]张军,赵卫民,郭文跃,等.苯并咪唑类缓蚀剂缓蚀性能的理论评价[J].物理化学学报,2008,24(7):1239-1244.(ZHANG Jun,ZHAO Weimin,GUO Wenyue, et al. Theoretical evaluation ofcorrosion inhibition performance of benzimidazole corrosion inhibitors[J]. Acta Physico-ChimicaSinica,2008,24(7):1239-1244.)
[10]卑凤利,陈海群,杨绪杰,等.2-氯甲基苯并咪唑的晶体结构、质子化特性及量子化学理论研究[J].化学学报,2004,62(3):328-333.(BI Fengli,CHEN Haiqun, YANG Xujie,et al.2-Chloromethyl-3H-benzoimidazol-1-ium nitrate:Crystal structure,protonation character and quantum chemical study[J].Acta Chimica Sinica, 2004,62(3):328-333.)
[11]BEREKET G,HUR E.Quantum chemical studies on some imidazole derivatives as corrosion inhibitors for iron in acidic medium[J].J MolecularStructure (Theochem),2002,578:79-88.
[12]张士国,杨频.用量子化学密度泛函理论研究环状含氮化合物分子结构与缓蚀性能的关系[J].中国腐蚀与防护学报,2004,24(4):240-244.(ZHANG Shiguo, YANG Pin. Themolecularstructure and corrosion inhibitor efficiency of some cyclic nitrogen compounds: An DFT study[J].Journal ofChinese Society for Corrosion and Protection,2004,24(4):240-244.)
[13]雷武,赵维,夏明珠.含硫缓蚀剂基于量子化学的定量构效-活性相关研究[J].计算机与应用化学,2003,20 (5):706-709.(L EI Wu,ZHAO Wei,XIA Mingzhu. QSAR studies of thiourea and tts derivaties based on the quantum calculation[J]. Computers and Applied Chemistry,2003,20(5):706-709.)
[14]CRUZJ,MARTINEZ-AGUILERA L M R,SALCEDO R, et al.Reactivity properties of derivatives of 2-imidaoline: An abinitio DFT study[J].International J Quantum Chemistry,2001,85(4):546-556.
