系列十六烷基邻二甲苯磺酸盐异构体的合成与结构表征
2010-01-29曲广淼程杰成李国桥
曲广淼,程杰成,丁 伟,于 涛,李国桥
(1.中国石油大学石油天然气工程学院,北京100220;2.大庆石油学院化学化工学院,黑龙江大庆163318; 3.大庆油田有限责任公司,黑龙江大庆163100)
烷基苯磺酸盐是目前三元复合驱矿场试验的主要驱油表面活性剂,研究烷基苯磺酸盐的结构与性能之间的关系对于提高油田采收率具有重要意义[1-2]。目前,驱油用的烷基苯磺酸盐大多是不同结构的烷基苯磺酸盐的混合物,很难确定表面活性剂分子结构与性能之间的关系。若想研究烷基苯磺酸盐的构效关系,必须要合成出各种类型、确定结构并且纯度高的烷基苯磺酸盐。Doe等[3-5]以苯为原料合成了一系列结构的十二烷基苯磺酸盐,并对其性能进行了研究。李宗石[6]、俞稼镛[7-8]、丁伟[2]等也分别合成出系列结构明确的烷基苯磺酸盐。笔者以邻二甲苯为原料,合成了一系列芳基在十六烷基链不同位置的十六烷基邻二甲苯异构体,并进一步合成了结构明确的十六烷基苯磺酸盐,为研究烷基苯磺酸盐的构效关系奠定了基础。
1 实验部分
1.1 药品与仪器
邻二甲苯、脂肪酰氯、α-溴代烷、无水三氯化铝、碘、镁带、无水乙醚、亚硫酸氢钠、水合肼、二缩三乙二醇、氯磺酸、高氯酸、氢氧化钠、冰醋酸均为分析纯试剂。Pd/C催化剂(质量分数5%),大连化学物理研究所提供。
辽阳市恒温仪器厂产低温浴槽,常州国华电器有限公司 G511型立式电动搅拌器,浙江舟山市定海区海源仪器厂 SHT型搅拌数显恒温电热套。海安科研仪器厂J X型真空泵,实验室自制高压反应釜,德国布鲁克光谱仪器公司Bruker-Tensor 27型傅里叶变换红外分光光度仪,英国安捷伦公司 SL型质谱仪,美国Varian公司Varian Unity-400型核磁共振波谱仪。
1.2 合成路线
采用邻二甲苯合成了芳基位于十六烷基长链1、2、3、4、5、6、7、8的8种十六烷基邻二甲苯同分异构体,再进行磺化反应,得到的产物为各种3,4-二甲基十六烷基苯磺酸盐,用代号 C16-noX表示,其中,C16代表十六烷基链,n为芳基在烷基碳链上的位置,o表示邻二甲苯基,X为 K、C、A和S,分别表示酮、醇、烃和磺酸盐。
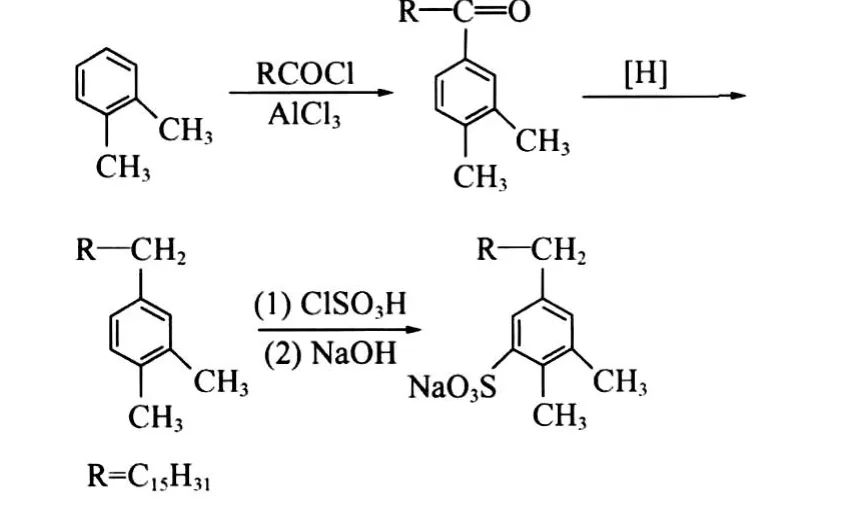
1.2.1 C16-1oS的合成路线
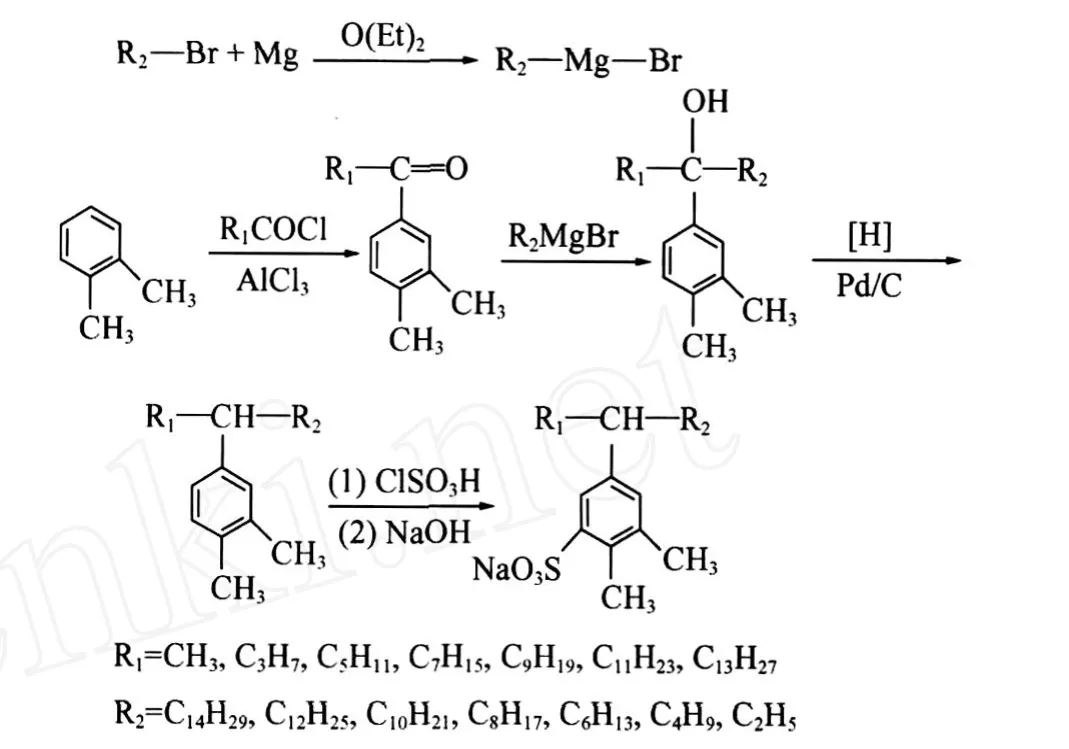
1.2.2 C16-2oS~C16-8oS的合成路线
1.3 3,4-二甲基十六烷基苯磺酸钠(C16-noS)的合成
8种3,4-二甲基十六烷基苯磺酸钠同分异构体的合成步骤相似,因此以C16-3oS的合成为例。
1.3.1 C16-noK的制备
在配有电动搅拌器、温度计、恒压滴液漏斗及回流冷凝管的反应瓶中,加入一定比例的邻二甲苯及无水AlCl3,搅拌,低温下缓慢滴加正十四酰氯。滴加完毕后,将反应体系升至一定温度,继续搅拌反应2 h。反应结束后,将反应混合物转移至冰稀盐酸溶液中使分层。取出油层并水洗至中性,再用无水CaCl2干燥,然后常压蒸除邻二甲苯,78 kPa下减压蒸馏收集310~317℃馏分,为C16-3oK。采用同样方法,分别滴加不同碳数的酰氯,制备出C16-1oK、C16-2oK、C16-4oK、C16-5oK、C16-6oK、C16-7oK、C16-8oK。
1.3.2 RMgBr(格氏试剂)的制备
在配有电动搅拌器、温度计及回流冷凝管的四口反应瓶中,N2保护下,加入12 g镁屑和少许碘,加热至碘成蒸气充满反应瓶,将45 g溴代乙烷和等体积乙醚缓慢滴加至反应瓶中,加热回流2 h后,冷却至室温,得到 C2H5MgBr。采用相同方法分别用溴代丁烷、溴代己烷、溴代辛烷、溴代癸烷、溴代十二烷、溴代十四烷制备其余7种格氏试剂。
1.3.3 C16-noC的制备
低温下向制得的格氏试剂体系中滴加 79 g C16-3oK和等体积无水乙醚的混合溶液,滴加完毕继续回流2 h,冷却至室温,将反应液倾入冰-稀盐酸混合液中使分层。依次用饱和NaHSO3溶液、饱和NaHCO3溶液和蒸馏水洗涤上层液体至中性,干燥后常压蒸出乙醚,减压蒸馏出沸程为312~315℃/ 78 kPa的馏分,即为C16-3oC。采用同样方法分别制备出 C16-2oC、C16-4oC、C16-5oC、C16-6oC、C16-7oC、C16-8oC。
1.3.4 C16-noA的制备
(1)C16-1oA的制备
向装有温度计、回流冷凝管、磁力搅拌器的反应瓶中,依次加入34.8 g C16-1oK、15.15 g水合肼及25 mL二缩三乙二醇,搅拌回流1 h。冷却至室温后安装分水器,加入16 g NaOH,加热蒸出水和过量的水合肼。温度升至190~200℃,继续反应2~3 h。冷却,将生成物转移至大烧杯中,加水稀释分层。分别用稀盐酸、蒸馏水洗涤有机相至中性,然后减压蒸馏,即得C16-1oA。
(2)C16-3oA的制备
将20 g C16-3oC、50 mL冰醋酸、1 mL高氯酸、1 g Pd/C催化剂(质量分数为5%)装入高压反应釜。密封后依次用N2、H2对高压反应釜进行置换,然后升温至60℃,向釜内充填 H2至1.0 MPa,磁力搅拌,保持釜内压力为1.0 MPa,直至压力恒定。用N2置换 H2后,开釜,滤去Pd/C催化剂。静止分层,用蒸馏水洗涤上层液体至中性,无水 CaCl2干燥,减压蒸馏收集304~306℃/78 kPa的馏分,即为C16-3oA。同样,分别以C16-2oC、C16-4oC、C16-5oC、C16-6oC、C16-7oC、C16-8oC为原料,制备出 C16-2oA、C16-4oA、C16-5oA、C16-6oA、C16-7oA、C16-8oA。
1.3.5 C16-noS的制备
在装有电动搅拌器、温度计、尾气吸收装置及恒压滴液漏斗的反应瓶中加入10.0 g C16-3oA,缓慢滴加3.7 g HSO3Cl,控制反应温度不超过20℃,滴加完毕继续搅拌2 h后停止反应。强烈搅拌下向体系加入质量分数20%的 NaOH水溶液进行中和。旋转蒸发除去溶剂,加入大量无水乙醇溶解、过滤,用沸程为60~90℃石油醚多次萃取,至石油醚层无色。取下层溶液旋转蒸发除去溶剂后,用乙醇重结晶分离,真空干燥即得 C16-3oS。同样分别以C16-1oA、C16-2oA、C16-4oA、C16-5oA、C16-6oA、C16-7oA、C16-8oA 为原料,制备出 C16-1oS、C16-2oS、C16-4oS、C16-5oS、C16-6oS、C16-7oS、C16-8oS。
2 结果与讨论
2.1 C16-3oK合成条件的优化
在合成 C16-3oK的过程中,为优化合成条件,采用正交实验考察了滴加温度、原料摩尔比和反应温度对 C16-3oK收率的影响。在条件实验基础之上,按照三因素四水平正交表 L9(34)设计了正交实验方案,正交实验的设计和实验结果分别列于表1和表2。
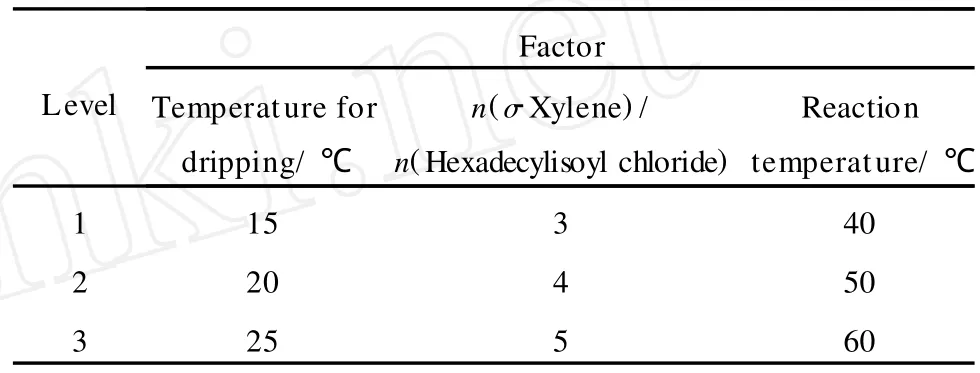
表1 C16-3oK合成条件优化的正交设计Table 1 Orthogonal design for condition optimization of C16-3oK synthesis
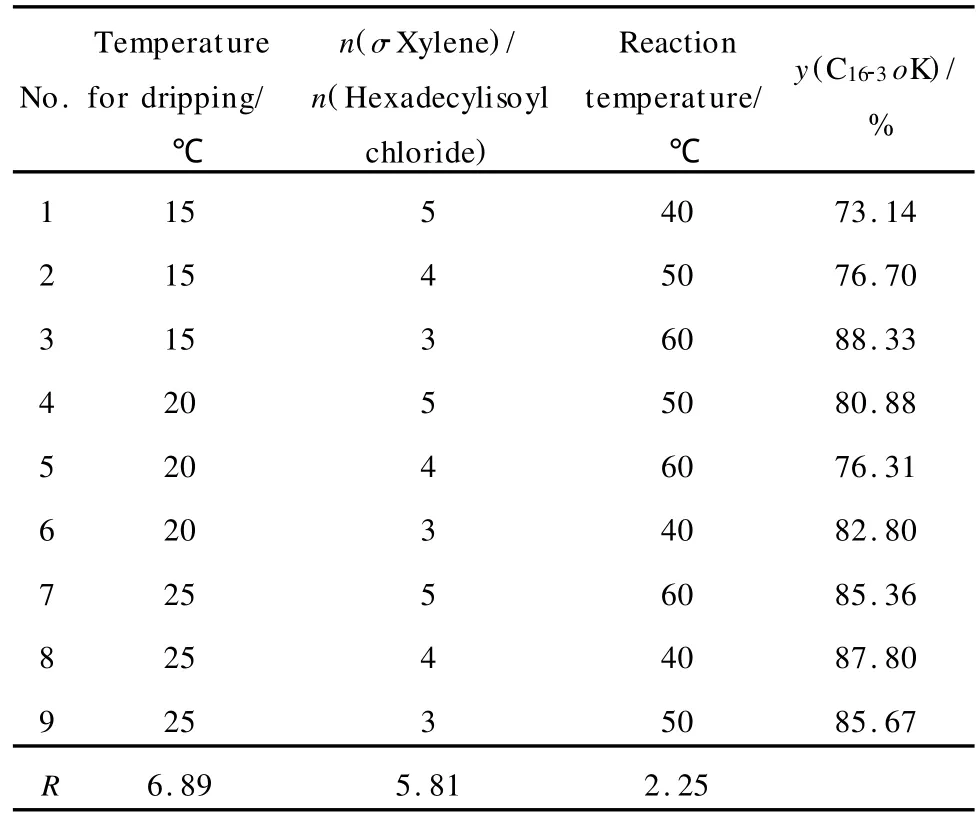
表2 C16-3oK合成条件优化的正交实验结果Table 2 Orthogonal experiment results for condition optimization of C16-3oKsynthesis
由表2可以看出,对 C16-3oK收率影响的主次顺序为滴加温度、邻二甲苯与酰氯的摩尔比和反应温度(极差分析分别为6.89、5.81和2.25),其中,滴加温度是影响该反应的主要因素。滴加温度为25℃、邻二甲苯与酰氯摩尔比为3、反应温度为60℃时,C16-3oK的收率最高,这就是合成 C16-3oK的最佳工艺条件。根据上述条件进行了平行实验,得到的产物收率在88.23%~90.63%。
2.2 中间体C16-noK、C16-noC、C16-noA的沸程
测得的中间体C16-noK、C16-noC、C16-noA的沸程列于表3。从表3可以看出,C16-noK的沸程随着其相对分子质量的增加而增加,C16-noC和 C16-noA的沸程无明显规律。

表3 C16-noK、C16-noC、C16-noA的沸程Table 3 Boiling ranges of C16-noK,C16-noC,C16-noA
2.3 C16-3oK、C16-3oC、C16-3oA和 C16-3oS的分子结构
由于所合成的中间体和产物在结构上相似,以C16-3oS为例对其中间体及最终产品的结构进行分析和表征。
2.3.1 C16-3oK的结构
图1为 C16-3oK的 FT-IR谱图。在 1000~1250 cm-1出现的峰为芳环上 C—H面内弯曲振动峰,1608和1453 cm-1的峰为苯环的 C—C伸缩振动峰,887和818 cm-1的峰为六元三取代芳环的特征峰,724 cm-1处为C—(CH2)n—C(n>4)的骨架振动峰,1683 cm-1处为的特征峰,在953和650 cm-1处未出现C—C—Cl和C—Cl的伸缩振动峰。
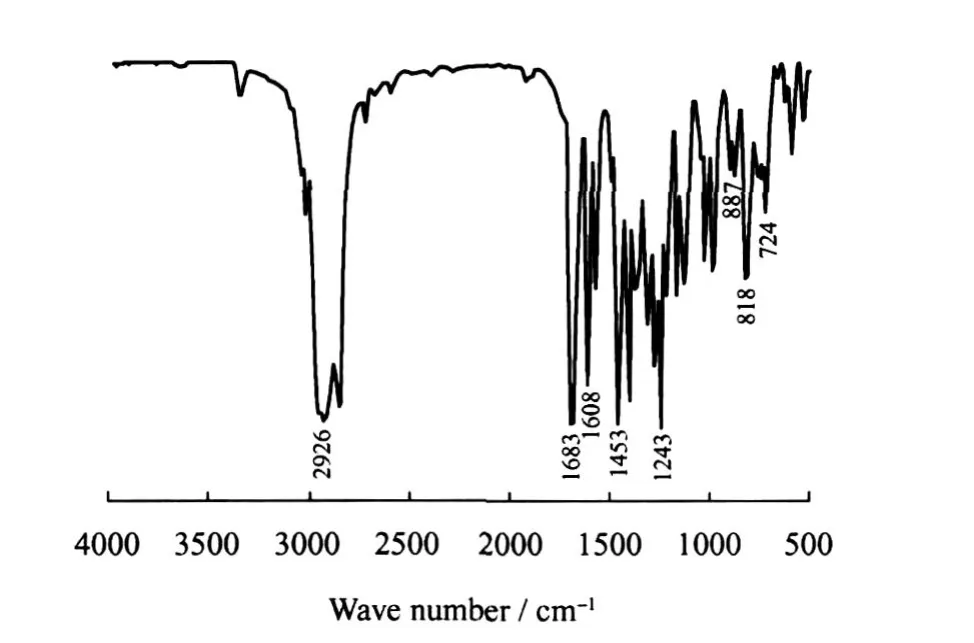
图1 C16-3oK的 FT-IR谱图Fig.1 FT-IR spectrum of C16-3oK
图2为 C16-3oK的1H NMR谱图(CDCl3, 300 MHz)。从图 2可得到,δ=0.854(a,3H), δ=1.065(b,2H),δ为1.145~1.918(c,d,18H), δ=1.918(e,2H),δ=2.279(f,2H),δ=3.397和δ=3.581(g,h,6H),δ为 7.261~7.569(i,j,k, 3H)。由此可知,所合成的化合物 C16-3oK有如图2所示的结构式。
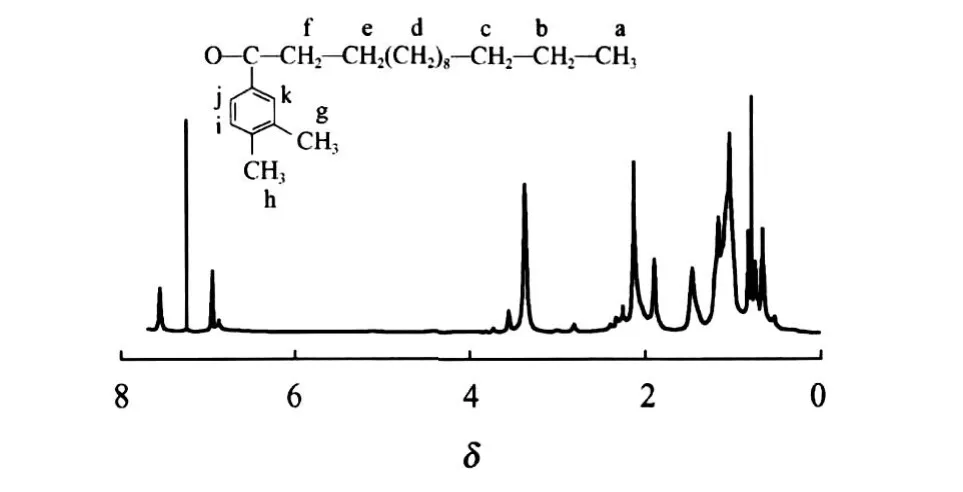
图2 C16-3oK的1H NMR谱图Fig.2 1H NMR profile of C16-3oK
2.3.2 C16-3oC的结构
图3为C16-3oC的 FT-IR谱图。在884、819 cm-1处的吸收峰为三取代芳环的特征峰,1567和1503 cm-1处为芳环的骨架振动峰,722 cm-1处为C—(CH2)n—C(n>4)的骨架振动峰,2924 cm-1处为直链烷烃 C—H伸缩振动峰,1378 cm-1处为—CH3的变形对称振动峰,3573 cm-1处为 O—H的伸缩振动吸收峰,1456 cm-1处为季碳醇的特征峰,且在1690 cm-1处没有出现的吸收峰,进一步说明C16-3oK与格氏试剂 C2H5MgBr反应生成了C16-3oC。
图4为C16-3oC的1H NMR谱图(CDCl3,300 MHz)。从图4可得到,δ为0.855~0.899(a,3H),δ为0.924~0.990(i,3H),δ为1.234~1.261(b~e, 22H),δ为 2.432~2.493(f,2H),δ为 1.754~1.778(h,2H),δ=5.699(g,1H),δ=6.913(l,1H), δ=7.052(m,1H),δ=7.056(n,1H),δ=2.150~2.289(j,k,6H)。由此可知,所合成的化合物C16-3oC有如图4所示的结构式。
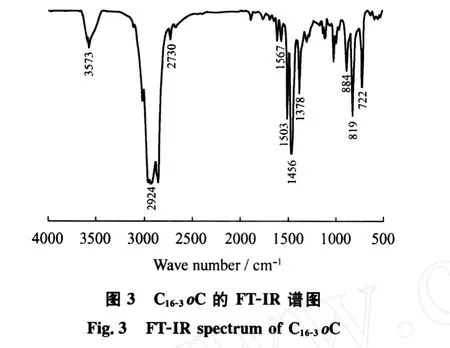
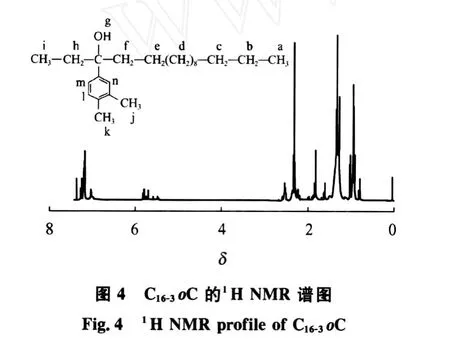
2.3.3 C16-3oA的结构
图5为 C16-3oA的 FT-IR谱。在884、819 cm-1处吸收峰为三取代芳环的特征峰,在1609、1507、1455 cm-1处为芳环的骨架振动峰;724 cm-1处为 C—(CH2)n—C(n>4)的骨架振动峰, 2925 cm-1处为直链烷烃 C—H 的伸缩振动峰, 1378 cm-1处为 CH3的变形对称振动峰,在3573 cm-1处未出现羟基特征峰,说明醇羟基已经被转化。
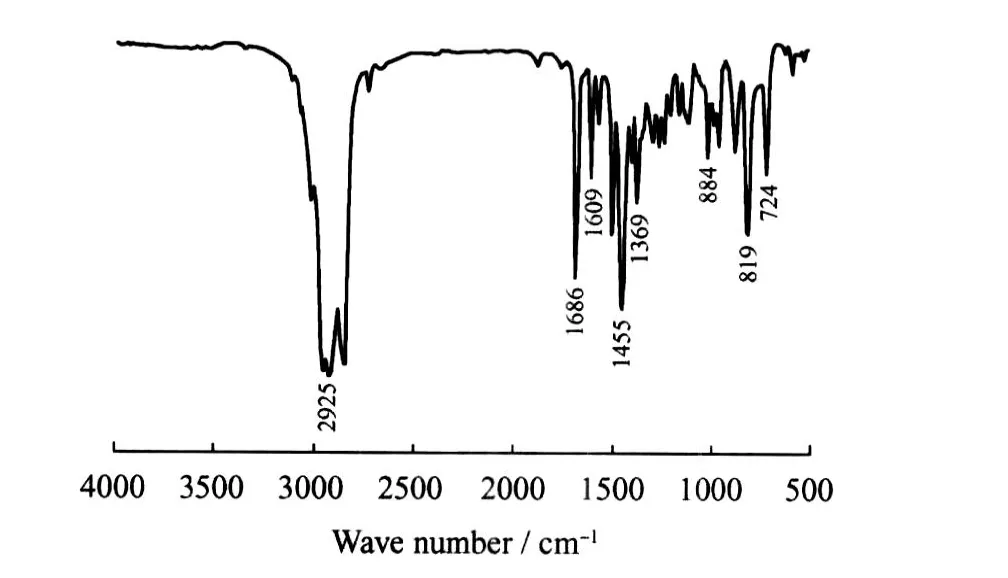
图5 C16-3oA的FT-IR谱图Fig.5 FT-IR spectrum of C16-3oA
图6为 C16-3oA 的1H NMR谱图(CDCl3, 300 MHz)。从图6可得到,δ在0.734~0.783(a, 3H),δ在0.855~0.899(i,3H),δ在1.206~1.298 (b~f,24H),δ在1.492~1.634(h,2H),δ=2.304 (g,1H),δ=6.887(l,1H),δ=7.024(m,1H),δ= 7.049(n,1H),δ在2.227~2.240(j,k,6H)。由此可知,所合成的化合物 C16-3oA有如图6所示的结构式。

图6 C16-3oA的1H NMR谱图Fig.6 1H NMR profile of C16-3oA
2.3.4 C16-3oS的结构
图7为C16-3oS的 ESI-MS谱图。图中只有1簇分子离子峰,没有其它杂质峰出现,说明产品中没有未磺化物和双磺化物。且单峰测定的相对分子质量为409,与分子设计中的 C16-3oS的阴离子相符。
图8为C16-3oS的FT-IR谱图。在1609、1559、1487 cm-1处为芳环的骨架振动峰;720 cm-1处为 C—(CH2)n—C(n>4)的骨架振动峰, 2925、2854 cm-1处为直链烷烃 C—H的伸缩振动峰,1378 cm-1处为 CH3的变形对称振动峰,在1203、1062、1020、879 cm-1处为特征峰,说明有磺酸基的存在。
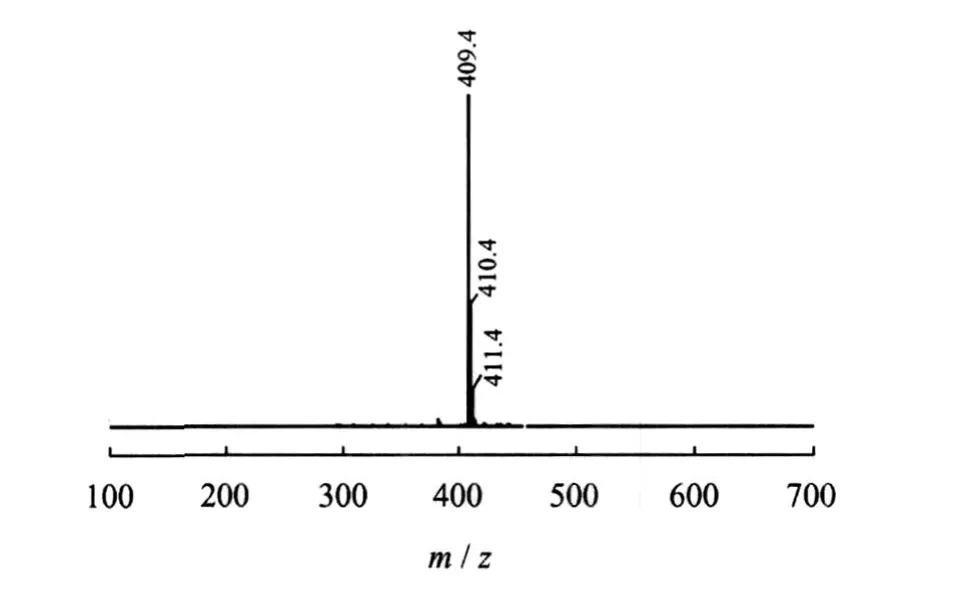
图7 C16-3oS的 ESI-MS谱图Fig.7 ESI-MS spectrum of C16-3oS

图8 C16-3oS的FT-IR谱图Fig.8 FT-IR spectrum of C16-3oS
图9为C16-3oS的1H NMR谱图(CDCl3,300 MHz)。从图9可得到,δ在0.637~0.685(a,3H),δ= 0.758(i,3H),δ在1.040~1.239(b~e,22H),δ= 1.492(h,f,4H),δ在 2.227~2.305(g,1H),δ= 7.567(m,2H),δ=6.948(n,3H),δ在 1.902~2.151(j,k,6H)。由此可知,所合成的化合物C16-3oS有如图9所示的结构式。
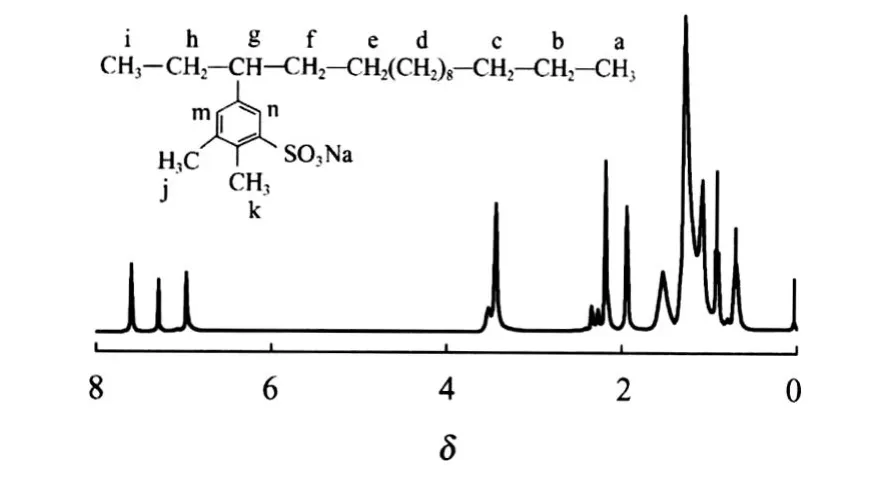
图9 C16-3oS的1H NMR谱图Fig.9 1H NMR profile of C16-3oS
3 结 论
(1)以邻二甲苯为原料,经 Friedel-Crafts酰基化反应、格氏反应、加氢还原反应等步骤合成出8种十六烷基邻二甲苯磺酸钠的异构体。
(2)通过正交试验确定了合成3,4-二甲基苯基十六烷基苯酮的最佳工艺条件,脂肪酰氯滴加温度25℃,邻二甲苯与酰氯的摩尔比为3∶1,反应温度60℃,在此条件下3,4-二甲基苯基十六烷基苯酮的收率达88.23%~90.63%。
(3)采用红外光谱、核磁共振氢谱及电喷雾质谱对十六烷基邻二甲苯磺酸钠合成中间体及产物的结构进行了表征,结果表明,所合成中间体和产物的分子结构与设计分子结构相同。
[1]程杰成.“十五”期间大庆油田三次采油技术的进步与下步攻关方向[J].大庆石油地质与开发,2006,25(1):81-86.(CHENG Jiecheng.Improvement and progress of Daqing oil field enhanced oil recovery technology during the fifth period[J].Geology and Exploration of Daqing Oil Field,2006,25(1):81-86.)
[2]丁伟,王瑞,于涛,等.2,5-二甲基十二烷基苯同分异构体的精细合成与表征[J].石油学报(石油加工),2007, 23(3):76-77.(DING Wei,WANG Rui,YU Tao,et al. Fine synthesis and characterization of 2, 5-dimethyldodecylbenzene isomers[J].Acta Petrolei Sinica (Petroleum Processing Section),2007,23(3):76-77.)
[3]DOE P H,EMARY M E L,WADE W H.Surfactants for producing low interfacial tensions I Linear alkylbenzenesulfonates[J].Journal ofAmerican Oil Chemist’s Society,1977,54(12):570-577.
[4]DOE P H,EMARY M E L,WADE W H.Surfactants for producing low interfacial tensions II Linear alkylbenzenesulfonates with additional alky substituents [J].Journal of American Oil Chemist′s Society,1978,55 (5):505-512.
[5]DOE P H,EMARY M E L,WADE W H.Surfactants for producing low interfacial tensions Ⅲ Di and trialkylbenzenesulfonates[J].Journal ofAmerican Oil Chemist′s Society,1978,55(55):513-519.
[6]李志刚,乔卫红,李宗石,等.烷基苯酮的合成[J].大连理工大学学报,2002,42(5):531-533.(LI Zhigang, QIAO Weihong,LI Zongshi,et al.Synthesis of alkyl phenones[J]. Journal of Dalian University of Technology,2002,42(5):532-535.)
[7]王琳,宫清涛,王东贤,等.支链烷基苯磺酸钠的合成、表征及其结构对表面性质的影响[J].石油化工,2004, 33(2):104-108.(WANG Lin,GONG Qingtao,WANG Dongxian,et al.Synthesis and characterization of sodium branched-alkylbenzene sulfonates[J]. Petrochemical Technology,2004,33(2):104-108.)
[8]姜小明,徐志成,史福强,等.高纯度支链三烷基苯磺酸钠的合成与表面活性[J].精细化工,2004,21(11):808-811.(J IANG Xiaoming,XU Zhicheng,SHI Fuqiang, et al. Synthesis and surface activity of pure sodium branched trialkylbenzenesulfonates[J].Fine Chemicals, 2004,21(11):808-811.)
