TiO2对 Ni-Mo/MCM-41催化剂加氢脱硫性能的影响
2010-01-29吴春雷王安杰
吴春雷,李 翔,2,王安杰,2
(1.大连理工大学精细化工国家重点实验室,辽宁大连116012;2.辽宁省高校石油化工技术与装备重点实验室,辽宁大连116012)
在传统的以γ-Al2O3作载体的双金属硫化物加氢脱硫(HDS)催化剂中,TiO2具有一定的助剂作用。它的引入一方面能够提高催化剂酸性[1],提高活性组分分散度[2]以及减弱活性组分与γ-Al2O3之间的相互作用,从而提高活性组分的可还原性和硫化能力[3];另一方面还起到了电子型助剂作用[4]。MCM-41介孔分子筛因其具有比表面积大、孔径均一、可调等特点,在大分子参与的非均相催化反应中展现出良好的应用前景[5]。笔者[6-7]前期研究结果表明,全硅MCM-41(Si-MCM-41)的硅质骨架结构使得它与活性组分间的相互作用适中,而其较大的比表面积和规整的一维介孔孔道则有利于活性组分分散和大分子含硫化合物的扩散,因而是一种优良的加氢脱硫催化剂载体。其担载制备的Co-Mo和Ni-Mo催化剂对于二苯并噻吩(DBT)的 HDS反应表现出了很高的催化活性。为了进一步提高Si-MCM-41担载的Ni-Mo催化剂的 HDS反应性能,以及探讨 TiO2的助剂作用,笔者采用分步浸渍的方法在 Ni-Mo/MCM-41催化剂中引入少量的TiO2,以DBT作为模型化合物,考察了 TiO2及其引入顺序对Ni-Mo/MCM-41催化剂加氢脱硫反应催化性能的影响。
1 实验部分
1.1 原料
钼酸铵((NH4)6Mo7O24·4H2O)、无水乙醇、钛酸四丁酯(C16H36O4Ti)和硝酸镍(Ni(NO3)2·6H2O)均为分析纯。五水偏硅酸钠(Na2SiO3·5H2O)为工业级。十六烷基三甲基溴化铵(CTAB)为南京旋光科技有限公司产品,工业级。DBT由联苯和硫合成[8]。十氢萘购自上海试剂分装厂,纯度>99%。
1.2 MCM-41的合成和催化剂的制备
按文献[9]方法制备Si-MCM-41。在Na2SiO3·5H2O中加入 H2SO4,调节p H值至11。按一定比例加入模板剂CTAB,在不锈钢压力釜中于120℃下晶化48 h。固体经过滤、洗涤、烘干后,于600℃和 N2保护下焙烧12 h,然后在空气中540℃焙烧6 h,得到Si-MCM-41。
采用共浸渍法制备 Si-MCM-41担载的 Ni-Mo催化剂。将计量的Si-MCM-41加入到钼酸铵和硝酸镍的水溶液中,在室温下浸渍8 h,所得固体产物在120℃下烘干12 h,在500℃空气中焙烧3 h,得到Ni-Mo/MCM-41催化剂。
采用分步浸渍的方法制备2种 TiO2改性的Ni-Mo/MCM-41催化剂。
为了得到 TiO2先于活性组分引入的催化剂,首先将Si-MCM-41加入到计量的钛酸四丁酯的无水乙醇溶液中,在室温下浸渍8 h,所得固体产物在120℃下烘干 12 h,500℃空气中焙烧 3 h,得到TiO2-MCM-41载体。然后将计量的 TiO2-MCM-41加入到钼酸铵和硝酸镍的水溶液中,在室温下浸渍8 h,120℃下烘干12 h,500℃空气中焙烧3 h,得到的催化剂记为Ni-Mo/TiO2-MCM-41。
为了得到在活性组分之后引入 TiO2的催化剂,将制备的Ni-Mo/MCM-41催化剂的前驱体加入到计量的钛酸四丁酯的无水乙醇溶液中,室温浸渍8 h后,经120℃烘干12 h,然后在500℃下焙烧3 h,得到的催化剂记为 TiO2-Ni-Mo/MCM-41。
在上述3种催化剂中,担载MoO3的质量分数均为20%,助剂Ni与Mo摩尔比为0.75,TiO2的质量分数为2%。
1.3 HDS反应
在内径为8 mm的不锈钢固定床中压反应器中进行 HDS反应。催化剂经压片,破碎至0.5~0.8 mm装填,用量0.1 g。HDS反应前,用含 H2S体积分数为10%的 H2S-H2混合气对氧化态催化剂进行硫化,硫化温度400℃,时间3 h。然后在压力4.0 MPa、氢/油体积比750、反应温度280℃的条件下进行 HDS反应。原料为质量分数0.8%的DBT/十氢萘溶液,反应停留时间(Reaction time,定义为催化剂质量(g)/反应物摩尔流量(mol/min))在12.5~50.3(g·min)/mol间变化。采用 Agilent 6890N型气相色谱仪,配有固定相为(5%)二苯基-(95%)二甲基聚硅氧烷的市售 HP-5型毛细管色谱柱,测定原料和反应产物的组成。
图1示出了DBT的 HDS反应网络[10]。DBT的 HDS反应主要通过直接脱硫(DDS)和预加氢脱硫(HYD)两条并行的反应路径进行。DDS反应路径的产物为联苯(BP),在有机硫化物如DBT存在的情况下,BP很难进一步加氢生成苯基环己烷(CHB)[11]。DBT的 HYD反应路径可以看作是1个串连反应,四氢硫芴 (TH-DBT)和六氢硫芴(HH-DBT)是 HYD反应路径的主要含硫中间体,脱硫后生成 CHB,再进一步加氢生成联环己烷(BCH)。在酸性较强的催化剂上,CHB和BCH还会发生加氢裂化反应,产物为苯(B)和环己烷(C)。在本研究所述实验条件下,没有检测到 TH-DBT、HH-DBT等含硫中间体,因此可以用DBT的转化率作为催化剂 HDS反应活性的指标。
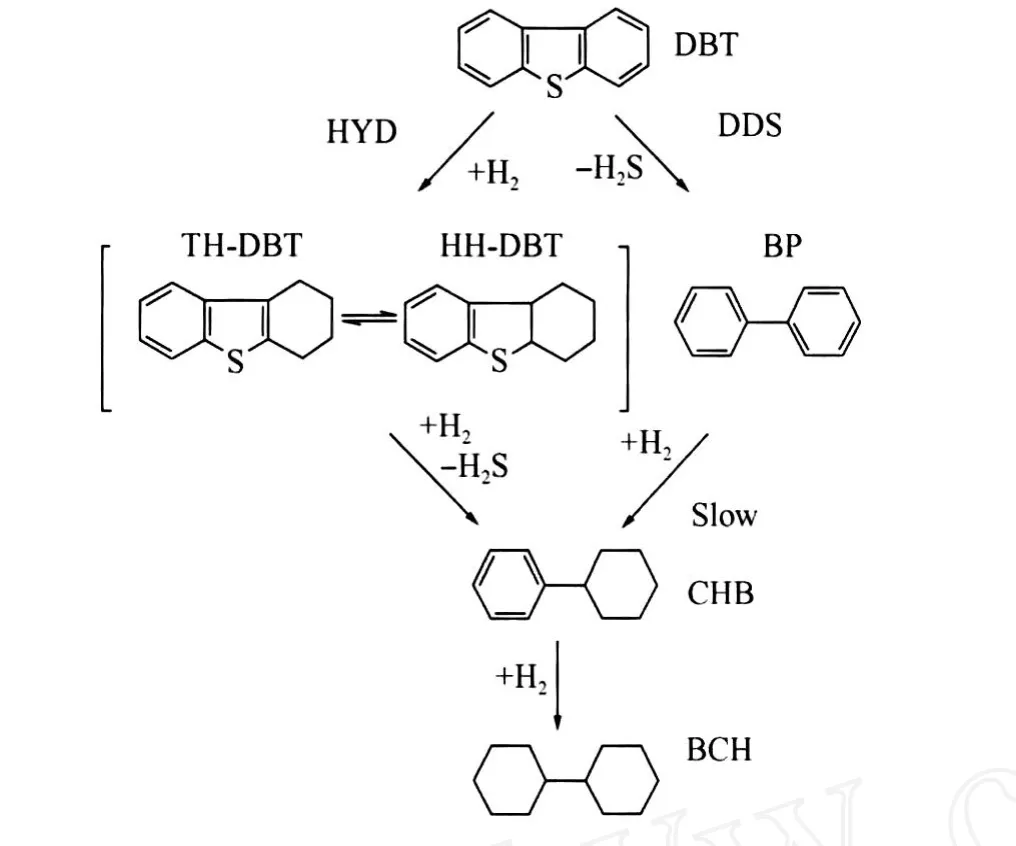
图1 二苯并噻吩(DBT)的 HDS反应网络Fig.1 HDS reaction network of dibenzothiophene(DBT)
1.4 催化剂表征
采用日本分光公司Jasco V-550型紫外/可见分光光度计测定催化剂前驱体的紫外/可见漫反射光谱(UV-Vis),扫描范围190~800 nm。在 Chembet-3000分析仪上进行 TPR试验。测试前,样品首先在 He气氛下200℃处理2 h。还原气为10%H2+ 90%Ar混合气(体积分数),升温速率10℃/min。在Chembet-3000分析仪上用 NH3-TPD测定催化剂前驱体的表面酸强度分布。将0.2 g样品放在U型石英玻璃管中,500℃下 He气吹扫0.5 h,然后降温至150℃并吸附 NH3至饱和,待基线平稳后以18℃/min速率升温至550℃,He流速20 mL/min。
2 结果与讨论
2.1 Ni-Mo/TiO2-MCM-41和 TiO2-Ni-Mo/MCM-41催化剂的表征结果
图2示出了 Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和 TiO2-Ni-Mo/MCM-41氧化物前驱体的UV-Vis谱图。在Ni-Mo/MCM-41氧化态前驱体的UV-Vis谱中,250和300 nm处的吸收峰分别对应四面体配位的 MoO2-4和八面体配位的 Mo6+物种[12-13],400~460 nm处的吸收峰对应八面体配位的 Ni2+物种[14]。Ni-Mo/TiO2-MCM-41和 Ni-Mo/ MCM-41的 UV-Vis曲线相差不大,说明 TiO2先于活性组分引入对Ni和Mo物种的配位状态影响不大。但TiO2-Ni-Mo/MCM-41与Ni-Mo/MCM-41的 UV-Vis相比,TiO2-Ni-Mo/MCM-41在400~460 nm处的紫外吸收峰强度降低,同时300 nm处的八面体配位的Mo6+物种的紫外吸收峰有所增强,说明 TiO2-Ni-Mo/MCM-41前驱体中八面体配位的Ni2+物种含量有所减少,而八面体配位的 Mo6+物种含量有所增加。
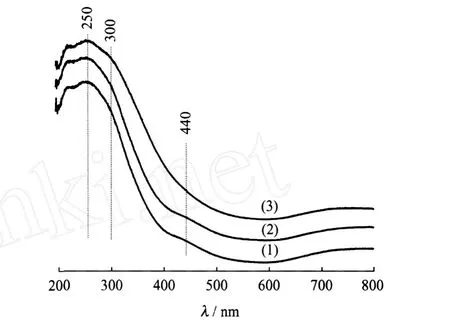
图2 Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和TiO2-Ni-Mo/MCM-41氧化物前驱体的 UV-Vis谱图Fig.2 UV-Vis patterns of Ni-Mo/MCM-41,Ni-Mo/ TiO2-MCM-41 and TiO2-Ni-Mo/MCM-41 oxidic precursors(1)Ni-Mo/MCM-41;(2)Ni-Mo/TiO2-MCM-41; (3)TiO2-Ni-Mo/MCM-41
图3为Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和 TiO2-Ni-Mo/MCM-41氧化态前驱体的 TPR谱图。Ni-Mo/MCM-41前驱体的 TPR谱图中主要有2个氢耗峰:403℃的低温氢耗峰归属为八面体配位的 Mo6+物种的部分还原(Mo6+→Mo4+)[15-16], 500℃左右的肩峰可能与α-NiMoO4相中 Ni物种的还原有关[17]。与 Ni-Mo/MCM-41相比,Ni-Mo/ TiO2-MCM-41的 TPR谱图中低温氢耗峰的还原温度和肩峰强度都有所降低,说明将 TiO2先于活性组分引入到MCM-41表面在一定程度上促进了前驱体的还原。这可能是因为 TiO2的引入增加了活性组分与载体之间的相互作用力,提高了活性组分的分散度[18-19]。而 TiO2-Ni-Mo/MCM-41的低温氢耗峰及其肩峰的还原温度分别比Ni-Mo/MCM-41相应氢耗峰的温度升高了5℃和23℃,在一定程度上抑制了催化剂中活性物种的还原。但总的说来,这3种催化剂的 UV-Vis和 TPR谱图差别并不明显,说明 TiO2的引入对Ni-Mo/MCM-41催化剂活性物种的配位状态和还原性能影响不显著。

图3 Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和TiO2-Ni-Mo/MCM-41氧化物前驱体TPR谱图Fig.3 TPR profiles of Ni-Mo/MCM-41,Ni-Mo/TiO2-MCM-41 and TiO2-Ni-Mo/MCM-41 oxidic precursors (1)Ni-Mo/MCM-41;(2)Ni-Mo/TiO2-MCM-41; (3)TiO2-Ni-Mo/MCM-41
图4为Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和TiO2-Ni-Mo/MCM-41氧化态前驱体的NH3-TPD谱图。根据 NH3的脱附温度,可将酸中心大致分为弱酸中心(150~250℃)、中强酸中心(250~350℃)和强酸中心(350~450℃)3类[20]。3种催化剂前驱体在230℃左右可以观察到明显的NH3脱附峰,可见3种催化剂前驱体表面主要为弱酸中心。此外,除 Ni-Mo/TiO2-MCM-41的 NH3脱附温度略有升高外,3者的 NH3-TPD谱图差别不大,说明引入 TiO2对Ni-Mo/MCM-41催化剂前驱体表面酸性影响不大。
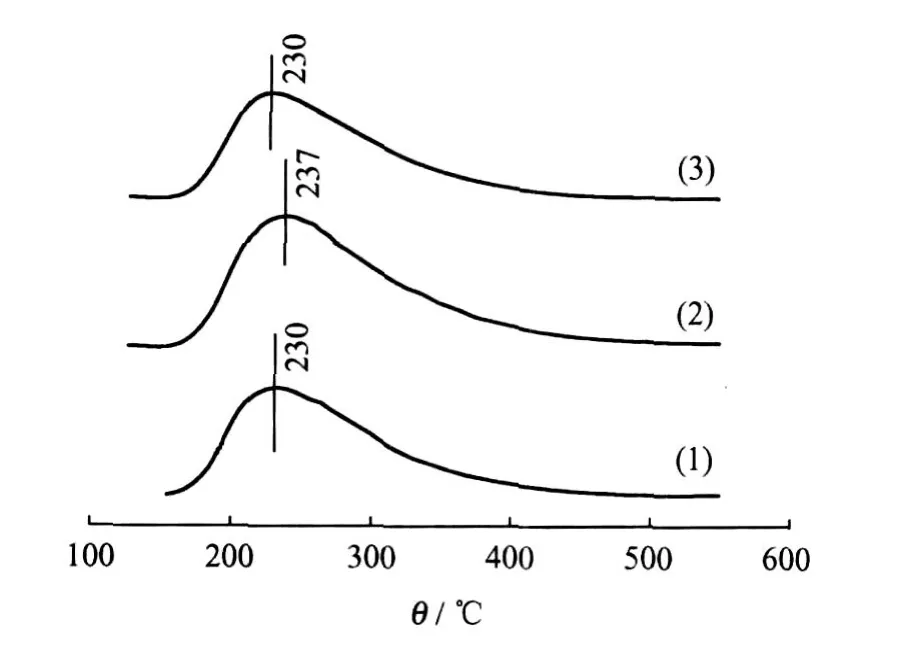
图4 Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和TiO2-Ni-Mo/MCM-41氧化物前驱体 NH3-TPD谱图Fig.4 NH3-TPD profiles of Ni-Mo/MCM-41,Ni-Mo/ TiO2-MCM-41 and TiO2-Ni-Mo/MCM-41 oxidic precursors(1)Ni-Mo/MCM-41;(2)Ni-Mo/TiO2-MCM-41; (3)TiO2-Ni-Mo/MCM-41
2.2 Ni-Mo/TiO2-MCM-41和 TiO2-Ni-Mo/MCM-41催化剂的HDS催化活性
图5示出了Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和 TiO2-Ni-Mo/MCM-41催化DBT HDS反应的产物组成随反应时间的变化。由图5可以看出,DBT在Ni-Mo/MCM-41催化剂上HDS反应的主要产物为BP,其次为CHB,另外还有少量的芳环完全加氢产物BCH和加氢裂化产物苯和环己烷(B+C)。而对于 TiO2改性的Ni-Mo/MCM-41催化剂,DBT在产物中的含量有不同程度的降低,转化率增加;从反应产物组成来看,DDS反应路径的产物BP含量有所降低,CHB和BCH等加氢反应路径产物尤其是BCH的量有了明显增加;在较长反应时间下, CHB甚至取代了BP成为主要的反应产物。另外, DBT在 TiO2改性的 Ni-Mo/MCM-41催化剂上进行 HDS反应时,加氢裂化产物苯和环己烷的量也明显提高。由于在有机硫化物如DBT存在的情况下,BP比较稳定,很难发生加氢裂化反应生成苯和/或环己烷[11],因此产物中苯和环己烷主要来源于加氢产物CHB和BCH的加氢裂化。从反应结果可以看出,TiO2的引入主要通过提高 Ni-Mo/ MCM-41催化剂 HYD反应路径的活性提高了其HDS活性。比较图5(2)和图5(3)可以看出,将TiO2引入到Ni-Mo/MCM-41前驱体的表面对催化剂HYD和 HDS活性的促进作用更为明显。
对于传统的以γ-Al2O3作载体的双金属硫化物HDS催化剂,TiO2主要通过提高催化剂酸性[1]、提高活性组分分散度[2]、减弱活性组分与γ-Al2O3之间的相互作用等几方面提高催化剂的 HDS反应活性[3]。另外,TiO2还起到了一定电子型助剂作用。在 HDS反应条件下,一部分 Ti4+可被部分还原为 Ti3+,Ti3+中3d轨道“富余”的电子注入 Mo的3d轨道后,使 Mo处于“富电子”状态,削弱Mo—S键,产生更多的硫空穴或配位状态不饱和的活性位,从而提高催化剂的活性[4]。但需要指出的是,目前广泛用于描述DBT直接脱硫过程的机理主要有氢解[21]和β-消除[22]两种。当催化剂表面处于富电子状态时,对氢解和β-消除反应都有促进,主要提高催化剂的DDS反应路径活性。这与本研究观察到的结果,即 TiO2主要通过促进催化剂 HYD反应路径的活性进而提高其 HDS反应活性不符。
根据 HDS反应产物的组成,TiO2改性的Ni-Mo/MCM-41催化剂表现出较高的加氢裂化活性,说明 TiO2的引入可能提高了Ni-Mo/MCM-41催化剂的酸性。但另一方面,图4中 NH3-TPD结果表明,不同催化剂氧化态前驱体表面酸性质差别不大,因此推测 TiO2引入更有可能促进“金属—SH”基团的生成,提高了硫化态催化剂的酸性。在酸性较强的催化剂上,活性中心能够通过与酸中心的相互作用形成“缺电子结构(Electron-deficient structure)”[23],这部分具有缺电子性质的活性中心同时具有较高的加氢活性。另外提高催化剂的酸性,也有利于氢的解离和高活性溢流氢物种的生成,从而提高催化剂的加氢活性[24-25]。与 Ni-Mo/TiO2-MCM-41相比,在 TiO2-Ni-Mo/MCM-41中可能有更多的 Ti物种暴露在催化剂表面,能更为有效地提高硫化物催化剂的酸性,因此 TiO2-Ni-Mo/ MCM-41表现出更高的 HYD活性和加氢裂化活性。
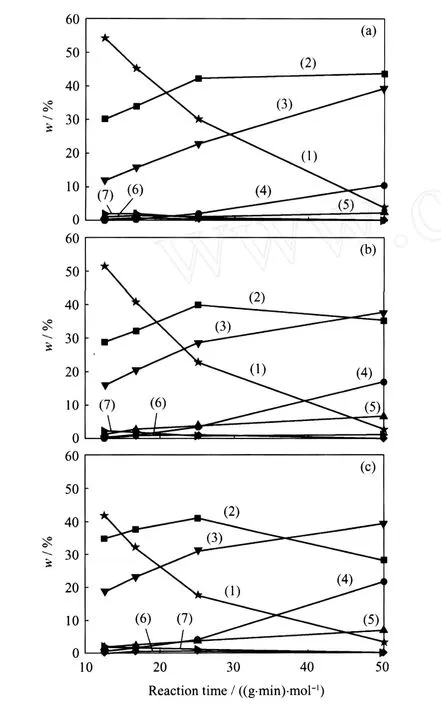
图5 Ni-Mo/MCM-41、Ni-Mo/TiO2-MCM-41和TiO2-Ni-Mo/MCM-41催化DBT HDS反应的产物组成随反应停留时间的变化Fig.5 Products compositions of DBT H DS over Ni-Mo/MCM-41, Ni-Mo/TiO2-MCM-41and TiO2-Ni-Mo/MCM-41 vs reaction time(a)Ni-Mo/MCM-41;(b)Ni-Mo/TiO2-MCM-41; (c)TiO2-Ni-Mo/MCM-41(1)DBT;(2)BP;(3)CHB;(4)B+C;(5)BCH; (6)HHDBT;(7)THDBTReaction time is specially appointed as the value of catalyst mass(g)/mol flow rate of reactant(mol/min)
3 结 论
(1)UV-Vis和 TPR结果表明,采用分步浸渍的方法将 TiO2先于 Ni-Mo活性组分引入对Ni-Mo/MCM-41前驱体中各物种的配位状态影响不大,但促进了其前驱体中各物种的还原;反之则减少了催化剂前驱体中八面体配位的 Ni2+物种的含量,但是提高了八面体配位的 Mo6+物种的含量,并在一定程度上抑制了活性物种的还原。NH3-TPD结果表明,Ni-Mo/MCM-41、TiO2-Ni-Mo/MCM-41以及Ni-Mo/TiO2-MCM-41 3种催化剂氧化态前驱体表面以弱酸中心为主。但总的说来,TiO2的引入对Ni-Mo/MCM-41催化剂氧化物前驱体的配位状态、还原性能以及酸性影响不显著。
(2)TiO2的引入虽然抑制了 Ni-Mo/MCM-41的直接脱硫活性,但是显著提高了其加氢反应路径的活性,进而提高了Ni-Mo/MCM-41总的 HDS反应活性。其中,将 TiO2在Ni-Mo活性组分之后引入能更有效地提高 Ni-Mo/MCM-41催化剂活性。根据反应产物组成分析,TiO2可能主要通过提高Ni-Mo/MCM-41硫化物酸性提高了催化剂的 HYD和HDS活性。
[1]RAMÍREZ J,RUÍZ-RAMÍREZ L,CEDENO L,et al. Titania-alumina mixed oxides as supports for molybdenum hydrotreating catalysts[J].Appl Catal:A, 1993,93(2):163-180.
[2]DAMYANOVA S,SPOJAKINA A,J IRATOVA K. Effect of mixed titania-alumina supports on the phase composition of NiMo/TiO2-Al2O3catalysts[J].Appl Catal:A,1995,125(2):257-269.
[3]邓存,周振华,童迅.TiO2调变对 MoO3/γ-Al2O3和CoO-MoO3/γ-Al2O3催化性能的影响[J].分子催化, 1998,12(2):107-112.(DENG Cun,ZHOU Zhenhua, TONG Xun.Effect of TiO2modifiedγ-Al2O3on the catalytic behavior of MoO3/γ-Al2O3and CoO-MoO3/ γ-Al2O3[J].Journal of Molecular Catalysis(China), 1998,12(2):107-112.)
[4]RAMÍREZA J,MACÍASA G,CEDEÑO L,et al.The role of titania in supported Mo,CoMo,NiMo and NiW hydrodesulfurization catalysts:An analysis of past and new evidences[J].Catalysis T oday,2004,98(1-2):19-30.
[5]YINGJ Y,MEHNERT C P,WONG M S.Synthesis and application of supramolecular templated mesoporous materials[J].Angew Chem Int Ed,1999,38(1-2):56-77.
[6]王瑶,王安杰,陈永英,等.以 MCM-41为载体担载Ni-Mo硫化物制备柴油深度加氢脱硫催化剂[J].石油学报(石油加工),2003,19(5):36-41.(WANG Yao, WANG Anjie,CHEN Yongying,et al.Preparation of Ni-Mo sulfide catalyst supported by MCM-41 for diesel deep hydrodesulfurization[J]. Acta PetroleiSinica (Petroleum Processing Section),2003,19(5):36-41.)
[7]王瑶,王安杰,陈永英,等.以 MCM-41为载体担载Co-Mo硫化物制备柴油深度加氢脱硫催化剂[J].石油学报(石油加工),2003,19(6):24-28.(WANG Yao, WANG Anjie,CHEN Yongying,et al.Preparation of Co-Mo sulfide catalyst supported by MCM-41 for diesel deep hydrodesulfurization[J]. Acta PetroleiSinica (Petroleum Processing Section),2003,19(6):24-28.)
[8]QIN W,ISHIHARA A,OGAWA S,et al.Study of hydrodesulfurization by the use of35S-labeled dibenzothiophene hydrodesulfurization mechanism on sulfided Mo/Al2O3[J].J Phys Chem,1994,98(3): 907-911.
[9]WANG A J,KABE T.Fine-tuning of pore size of MCM-41 by adjusting the initial p H of the synthesis mixture[J].Chem Commun,1999,20:2067-2068.
[10]HOUALLA M,NAG N K,SAPRE A V,et al. Hydrodesulfurization of dibenzothiophene catalyzed by sulfided CoO-MoO3/γ-Al2O3:The reaction network[J]. AICh E J,1978,24(6):1015-1021.
[11]REN J,WANG A J,LI X,et al.Hydrodesulfurization of dibenzothiophene catalyzed by Ni-Mo sulfides supported on a mixture of MCM-41 and HY zeolite[J]. Appl Catal:A,2008,344(1-2):175-182.
[12]ARNOLDY P,FRANKEN M C,SCHEFFER B,et al. Temperature-programmed reduction of CoO-MoO3/ A12O3catalysts[J].J Catal,1985,96(2):381-395.
[13]RAMÍREZ J,CASTILLO P,CEDEÑO L,et al.Effect of boron addition on the activity and selectivity of hydrotreating CoMo/Al2O3catalysts[J].Appl Catal: A,1995,132(2):317-334.
[14]建谋,杨先春,石亚华,等.氟在γ-Al2O3及 NiW/ γ-Al2O3催化剂中的作用 Ⅱ镍和钨的状态[J].分子催化,1990,4(3):181-187.(J IAN Mou,YANG Xianchun,SHI Yahua,et al.Effects of fluoride inγ-Al2O3and NiW/γ-Al2O3catalysts Ⅱ The states of nickel and tungsten[J].Journal of Molecular Catalysis (China),1990,4(3):181-187.)
[15]HENKER M,WENDLANDT K P,VAL YON J,et al. Structure ofMoO3/Al2O3-SiO2catalysts[J]. Appl Catal,1991,69(1):205-220.
[16]RAMÍREZJ,CONTRERAS R,CASTILLO P. Characterization and catalytic activity of CoMo HDS catalysts supported onalumina-MCM-41[J].Appl Catal:A,2000,197(1):69-78.
[17]BRITO JL,BARBOSA A L. Effectofphase composition of the oxidic precursor on the HDS activity of the sulfided molybdates of Fe(II),Co(II),and Ni(II) [J].J Catal,1997,171(2):467-475.
[18]RAN M S,MAITY S K,ANCHEYTA J,et al.TiO2-SiO2supported hydrotreating catalysts: Physicochemical characterization and activities[J].Appl Catal: A,2003,253(1):165-176.
[19]KLIMOVA T,RODRÍGUEZ E,MARTÍNEZ M,et al. Synthesis and characterization of hydrotreating Mo catalysts supported on titania-modified MCM-41[J].Microporous and Mesoporous Materials,2001,44-45:357-365.
[20]RYNKOWSKI J M,PARYJCZAK T,L ENIK M.On the nature of oxidic nickel phases in NiO/γ-Al2O3catalysts[J].Appl Catal:A,1993,106(1):73-82.
[21]TODOROVA T,PRINS R,WEBER T.A density functional theory study of the hydrogenolysis reaction of CH3SH to CH4on the catalytically active(100)edge of 2H-MoS2[J].J Catal,2005,236(2):190-204.
[22]BATAILL E F,L EMBERTEN J L,MICHAUD P, et al.Alkyldibenzothiophenes hydrodesulfurization——Promoter effect,reactivity and reaction mechanism[J]. J Catal,2000,191(2):409-422.
[23]ZENG S Q,BLANCHARD J,BREYSSE M,et al. Mesoporous materials from zeolite seeds as supports for nickel–tungsten sulfide active phases Part 2 Catalytic properties for deep hydrodesulfurization reactions[J]. Appl Catal:A,2006,298(1):88-93.
[24]AMBS W J,MITCHELL Jr M M.Hydrogen spillover on platinum-alumina,effect of water[J].J Catal,1983, 82(1):226-229.
[25]MILLER J T,MEYERS B L,MODICA F S,et al. Hydrogen temperature-programmed desorption (H2TPD)of supported platinum catalysts[J].J Catal, 1993,143(2):395-408.
