基于高通量测序的抑郁症患者肠道菌群结构分析
2023-12-24王雅萍潘苗苗夏欣媛肖振明杨浩吴志国赵超
王雅萍,潘苗苗,夏欣媛,肖振明,杨浩,吴志国,赵超,4
1. 上海市嘉定区南翔医院 心理医学科,上海 201800; 2. 复旦大学基础医学院教育部医学分子病毒学重点实验室, 上海 200032; 3. 上海市杨浦区精神卫生中心科教科,上海健康医学院精神卫生临床研究中心,上海 200433; 4. 国家老年疾病临床医学研究中心(复旦大学附属华山医院),上海 200040
重性抑郁障碍(major depressive disorder,MDD)也被称为临床抑郁症,是一种常见的慢性、致残性精神障碍[1]。全球有超过3.5亿人罹患抑郁症,造成了严重的医疗卫生和经济负担[2]。高度复杂的遗传差异和环境因素共同导致了抑郁症的高复发性和易感性[2],同时也导致现有医疗手段治疗效果的有限性。不少研究已关注到肠道菌群平衡在维护宿主健康中发挥了重要的作用[3-4]。近年来有研究证实肠道微生物能够通过多种途径调节中枢神经系统的发育,而肠道菌群失调与多种神经精神疾病有很高的相关性[5]。因此,本研究通过16S rRNA高通量测序,对抑郁症患者与健康人的粪便样本进行对比分析,并基于抑郁症患者的肠道菌群结构,为抑郁症的治疗干预提供潜在的微生物靶点。
1 材料与方法
1.1 研究对象
抑郁症患者组为2020年5月—2021年2月上海市嘉定区瑞金医院南翔分院门诊就诊的患者,共19例(男性9例,女性10例),年龄22~40岁。研究队列的入组标准为:①符合美国《精神障碍诊断与统计手册》(第五版DSM-5)“重性抑郁障碍”诊断标准,由2名主治医师以上职称的精神科医师诊断一致;②无胃肠道、高血压、糖尿病等系统性疾病,无双相情感障碍等其他精神障碍;③粪便标本收集前3个月无抗生素、益生元等微生态制剂使用史。健康对照组为同期参与体检的健康志愿者,共20名(男性10名,女性10名),年龄24~40岁。入组标准:①无精神疾病及家族史,无精神发育迟滞/障碍;②无重大躯体疾病,不合并胃肠道、高血压、糖尿病等系统性疾病;③粪便标本收集前3个月无抗生素、益生元等微生态制剂使用史。参与者均自愿参与并签署知情同意书。本研究经所在医院伦理委员会批准同意执行(伦理编号2020-6)。
1.2 粪便样本收集与肠道微生物DNA提取
在清晨未进食前,使用一次性15 mL无菌采便管收集患者和健康人的粪便标本2~3 g,于-80 ℃冰箱内保存。使用OMEGA DNA试剂盒(Omega Bio-Tek, Norcross,美国),按照制造商的说明书提取总基因组DNA样本,并在进一步分析之前储存在-20 ℃冰箱中。使用NanoDrop NC2000分光光度计(美国Thermo Fisher Scientific)和琼脂糖凝胶电泳来评估提取的DNA样品的数量和质量。
1.3 量表评估
告知所有参与者问卷调查的目的和具体要求,并进行抑郁症自我评估量表(PHQ-9)、广泛性焦虑障碍量表(GAD-7)评估和一般情况调查表评估。①PHQ-9用于筛查及评估抑郁症状,共有9个条目,总分值越高则抑郁程度越严重;②GAD-7为简明的焦虑症状自评表,共7个条目,总分值越高则焦虑程度越严重;③一般情况调查表主要包括对年龄、性别、家族有无精神疾病史、既往病史和现病史、失眠及便秘等情况的调查。
1.4 聚合酶链反应扩增及测序
使用正向引物338F(5′-ACTCCTACGGGAG-GCAGCA-3′)和反向引物806R(5′-GGACTA-CHVGGGTWTCTAAT-3′)对细菌16S rRNA基因V3-V4区域进行聚合酶链反应(polymerase chain reaction, PCR)扩增。PCR体系包含 5 μL 缓冲液(5×),0.25 μL快速pfu DNA聚合酶(5 U/μL),2 μL(2.5 mmol/L)dNTPs,1 μL(10 μmol/L)正向和反向引物,1 μL DNA模板和14.75 μL ddH2O。反应条件为98 ℃初始变性5 min,98 ℃变性30 s、53 ℃退火30 s、72 ℃延伸45 s的25个循环,最后在72 ℃延伸5 min。用Vazyme VAHTSTM DNA清洁珠(南京Vazyme)纯化PCR扩增子,并使用Quant-iT PicoGreen dsDNA检测试剂盒(美国Invitrogen)进行定量。扩增子经量化汇集后,使用Illumina NovaSeq平台和NovaSeq 6000 SP试剂盒(500个循环)进行成对端2×250 bp测序。
1.5 生物信息学分析与统计
使用DADA2进行引物去除、质量过滤、拼接和嵌合体去除,并使用QIIME2进行评估。使用Vsearch(v2.13.4_linux_x86_64)在97%的相似度水平上对测序读数进行映射。参照Greengenes数据库(Release 13.8, http://greengenes.secondgenome.com/)对获得的运算分类单元(operational taxonomic unit, OTU)数据进行分析。使用QIIME2调用mafft和FastTree构建系统发育树。在OTU水平上使用了Chao1指数、ACE指数、Shannon指数和Simpson指数分析物种Alpha多样性。使用Unifrac距离进行主坐标分析。使用QIIME2统计特征表来分析门和属水平的物种组成。使用线性判别分析(LEfSe)来分析样本间存在的差异微生物(Kruskal-WallisP值小于0.05,LDA得分阈值大于2.0)。R(4.0.0版)用于心理量表和样品的OTU丰度的Pearson相关分析及可视化。使用SPSS 16.0 进行数据分析,采用Kolmogorov-Smirnov来检验数据的正态性,采用t检验和Wilcoxon检验来评价数据差异的显著性,双侧P<0.05代表差异具有统计学意义。
2 结果
2.1 一般情况统计
健康组与抑郁症组男女比例均接近1∶1,抑郁症组年龄为29.00±8.09岁,健康对照组年龄为34.65±4.44岁。抑郁症组出现失眠和便秘情况的比例分别达到了79.0%和57.9%,而健康组失眠和便秘比例分别为10%和15%,两组失眠与便秘状况的构成比差异具有统计学意义(P<0.05)(见表1)。
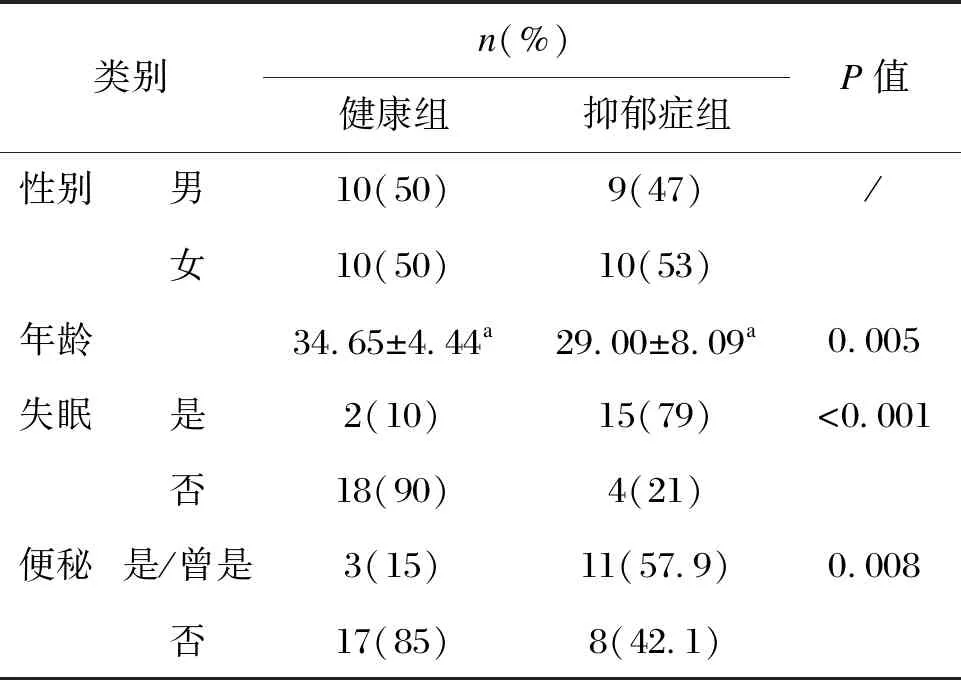
表1 一般情况统计
2.2 微生物结构分析
2.2.1 肠道菌群多样性分析Alpha多样性稀释曲线显示,两组样本的测序量都达到了饱和,多样性指标趋于稳定(见图1A)。抑郁症组与健康对照组Chao1指数与ACE指数的差异无统计学意义(P=0.547 5,P=0.493 8),即两组肠道微生物样本的群落丰富度无显著差异。两组Shannon指数与Simpson指数的差异也无统计学意义,表明两组被试肠道微生物群落的多样性没有显著差异(见表2)。使用Unifrac距离算法对比两组样本的Beta多样性,健康组样本分布较为集中,抑郁症组样本较为分散,两组样本点重合度较低,提示了两组被试肠道微生物群落构成存在差异(见图1B)。
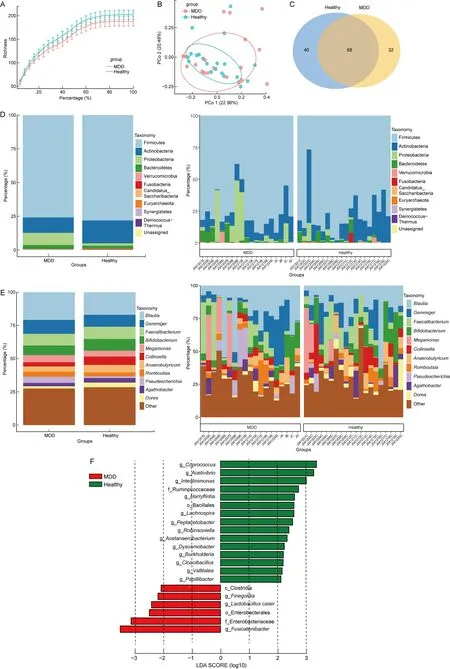
A: Alpha多样性稀释曲线,红色为抑郁症组,蓝色为健康组;B: Beta多样性主坐标分析(PCoA);C: OUT聚类维恩图;D~E: 门/属水平微生物堆积柱状图;F: LEfSe差异微生物群的LDA柱状图。
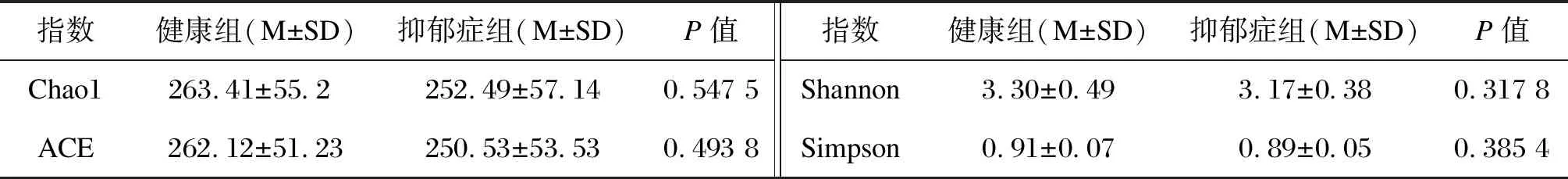
表2 肠道微生物Alpha多样性
2.2.2 肠道菌群物种组成分析聚类分析显示,健康对照组和患者组样本中检测到68个共有OUTs(见图1C)。健康组与抑郁症组在门水平结构组成相似,主要为厚壁菌门(Firmicutes)、放线菌门(Actinobacteria)、变形菌门(Proteobacteria)和拟杆菌门(Bacteroidetes)(见图1D)。与抑郁症组相比,健康组厚壁菌门和放线菌门的相对丰度更高,而变形菌门和拟杆菌门的相对丰度较低。两组样本在属水平的物种组成上也体现出明显差异。样本丰度均值比较显示,抑郁症组布劳特氏菌属(Blautia)、芽殖菌属(Gemmiger)、巨单胞菌属(Megamonas)和埃希菌属(Pseudescherichia)明显高于健康组(见图1E)。LEfSe分析展示了两组样本中具有显著差异的标志性物种,以LDA>2为阈值在健康组中共筛选到15个差异微生物类别,分别为粪球菌属(Coprococcus),醋弧菌属(Acetivibrio),Int~estinimonas,瘤胃球菌科(Ruminpcoccaceae),Harryflintia,芽孢杆菌目(Bacillales),毛螺菌属(Lachnospira),Peptacetobacter,罗宾逊贝属(Robinsonella),Acetanaerobacterium,韦氏非渗透杆菌属(Dysosmobacter),伯克霍尔德氏菌属(Burkholderia),Cloacibacillus,Vallitalea和Paoillibacter。在抑郁症组中共筛选到6个标志性物种,分别为大芬戈尔德菌属(Finegoldia), 干酪乳杆菌 (Lactobacilluscasei), 肠杆菌目(Enter~obacterales),肠杆菌科(Enterobacteriaceae), 纺锤链杆菌属(Fusicatenilbacter),和梭状芽孢杆菌(Clostridia)(见图1F)。
2.3 抑郁量表与微生物组相关分析
在PHQ-9和GAD-7总分的比较中,抑郁症组和健康组得分的差异有统计学意义(P<0.001,见图2A)。为调查肠道微生物与心理健康状况的相关性,结合两组具有显著差异的18种微生物类别与心理评估量表总分进行Pearson相关分析,可知健康组富集到的类球菌属(Coprococcus)与GAD-7总分呈显著负相关(P<0.05),Peptacetobacter与PHQ-9总分存在显著正相关(P<0.05,见图2B),其余差异微生物与两种量表分别呈现出不同的相关性,但不具有统计学意义。

A: 抑郁症组与健康组的PHQ-9和GAD-7量表总分直方图,***P<0.001;B: PHQ-9和GAD-7总分与LEfSe差异微生物的Pearson相关性分析,*P<0.05。
3 讨论
为尽可能减少影响菌群的变量因素的干扰,本研究以菌群结构相对稳定的青年人(20~45岁)为研究对象,限于嘉定地区,并排除了躯体疾病和抗生素、益生菌等对菌群的干扰进行研究。本研究共收集到19例抑郁症患者和20例健康人的粪便样本,经16S rRNA高通量测序和生物信息学分析显示,与健康人相比,抑郁症患者存在肠道菌群失衡。抑郁症组与健康组的微生物α多样性的差异无统计学意义,而β多样性体现出了明显的分群(PC1解释度为22.96%,PC2解释度为20.49%)。菌群物种组成分析表明,两组样本在门和属分类水平的构成和相对丰度均存在差异,抑郁症组的肠道菌群结构和组成均出现了显著的变化。进一步的标志性物种分析显示,抑郁症组的粪便样本中富集了肠杆菌科(Enterobacteriaceae)[6]和大芬戈尔德菌属(Finegoldia)[7]等炎症相关的微生物。炎症激活被认为是抑郁症和焦虑症发病机制中重要的一环,重性抑郁障碍患者在急性期促炎因子水平较高[8],而肠道微生物群的特异性变化以及由此产生的促炎级联反应可能诱发了抑郁症的免疫失调[9],在疾病发生发展中具有重要作用。此外,全身系统性炎症反应或外周炎症信号可以通过血脑屏障传递进入中枢神经系统[10],细菌代谢物的浸润也会直接或间接诱导中枢神经系统的炎症激活,进而造成肠脑轴的功能失调[11],可成为精神疾病的病理生理学基础。因此,肠道微生物群的失衡可能参与了抑郁症的发病机制,针对特异微生物靶点的干预或许可以有效控制抑郁症患者体内的炎性状态。
本研究在对心理健康状况与标志性微生物的相关分析中发现,健康组富集的粪球菌属(Coprococcus)与焦虑状况呈显著负相关,抑郁症患者样本组中并未富集到粪球菌,这与近期研究的结论相似[12]。有研究报道,抑郁症患者在接受治疗后,其粪便中粪球菌的比例显著增高[13],而粪球菌的丰度与良好的临床治疗表现明显正相关[14],因此粪球菌的相对丰度或可作为抑郁症临床转归的微生物预测指标。其他类似研究多以自然队列为研究对象,然而影响菌群的变量较多,导致难以发现与抑郁症本身相关的菌群特征。本研究尽可能排除影响菌群的潜在混杂因素,从而更容易获得与抑郁症本身相关的菌群结构特征。此外,由于研究样本量有限,且抑郁症样本组存在便秘的患者比例较高,因此这种基线差异可能对健康人与抑郁症患者肠道微生物状态的比较产生潜在影响。综上所述,本研究发现了肠道微生物结构失调与抑郁症的相关性,并提出了治疗抑郁症的潜在微生物靶点,为抑郁症的治疗和干预提供了相应的依据。
