基于审评的视角对呼吸湿化治疗用相关设备常见风险点及注册问题研究
2023-11-06陈虹蓁吴林蔚赖锦坝陈钿广东省药品监督管理局审评认证中心广东广州510080
陈虹蓁 吴林蔚 赖锦坝 陈钿 广东省药品监督管理局审评认证中心 (广东 广州 510080)
内容提要: 对呼吸湿化治疗用相关设备的常见风险点进行罗列分析,并从技术审评的角度对该类产品在注册申报过程中的问题提出建议,以期为生产企业与注册审评人员提供参考。
在开展医疗器械技术审评时,需根据申报资料把握产品安全性、有效性、可靠性并对其作出综合评价。而作为医疗器械注册申报资料中的重要组成部分,医疗器械风险管理资料需要能够完整涵盖器械技术风险管理要素并进行技术风险分析,企业应通过风险分析、风险评价、风险控制、剩余风险评价,最终输出风险管理报告,确保综合剩余风险在可接受的范围之内。新型冠状病毒肺炎疫情时期,呼吸湿化治疗用相关设备临床需求量骤增,越来越多的企业申报注册该类设备,但该产品在生产环节、投入临床使用时均存在一定的风险,如果不加强风险管理,可能会导致不良事件的发生,严重的甚至会危及患者生命安全。因此,本文对呼吸湿化治疗用相关设备的常见风险点进行罗列分析,并从技术审评的角度,对该类产品在注册申报过程中的问题提出建议,以期为生产企业与注册审评人员提供参考。
1.呼吸湿化治疗用相关设备常见风险点
目前我国现行有效的医疗器械风险管理标准为YY/T 0316-2016《医疗器械 风险管理对医疗器械的应用》[1],等同采用ISO 14971:2007更正版。欧盟MDR附录Ⅰ第一章给出了风险管理的详细要求。由于GB/T 42062-2022《医疗器械 风险管理对医疗器械的应用》将在2023年11月1日实施。
本文主要根据GB/T 42062《医疗器械 风险管理对医疗器械的应用》[2],对呼吸湿化治疗相关设备(如呼吸湿化治疗仪、空氧混合器、医用呼吸道湿化器及其配套使用的附件等)的已知或可预见的风险进行判定,产品在进行风险分析时至少应包括以下的主要风险点(详见表1)。
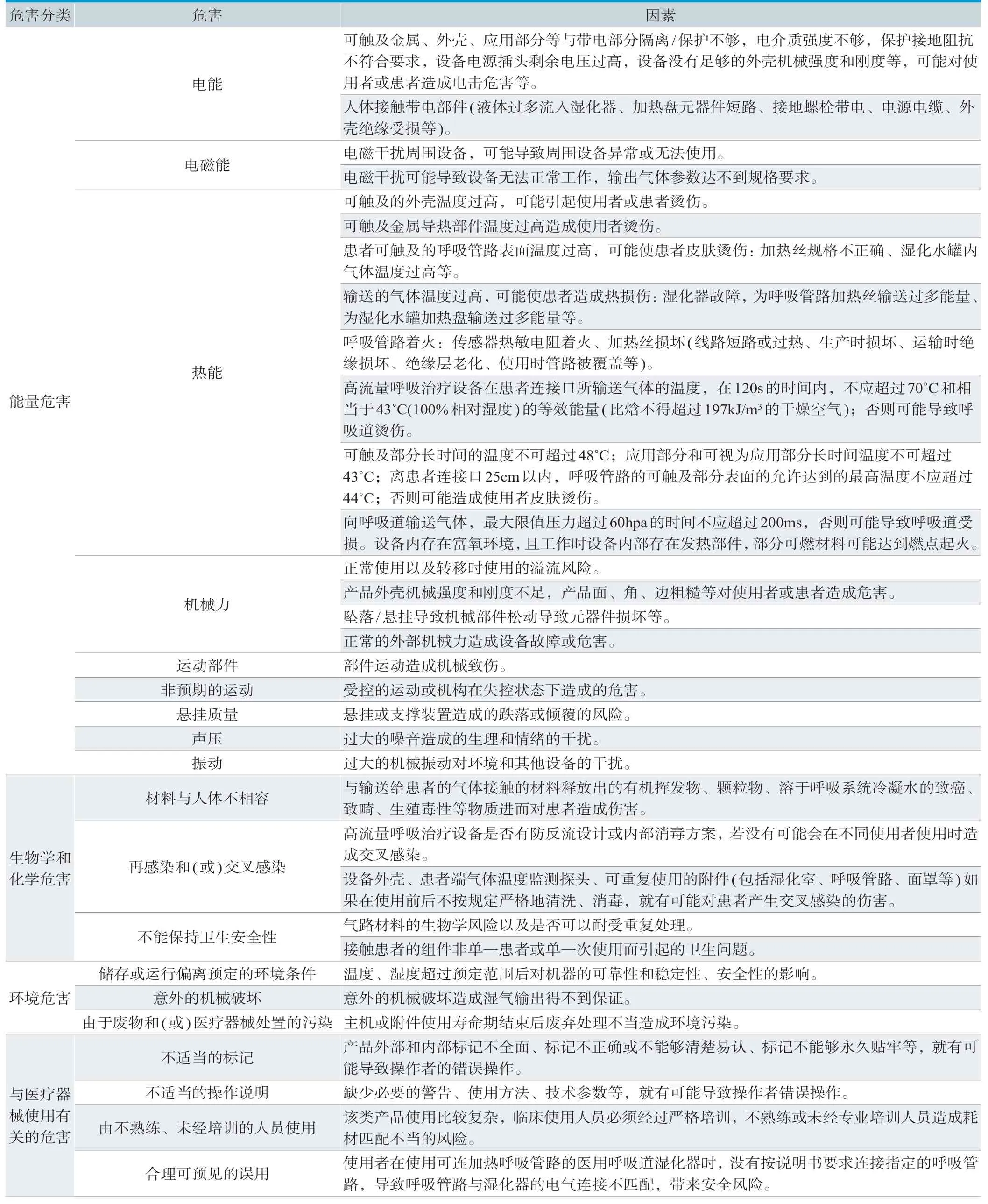
表1. 产品主要危害因素
表1所列出的风险点、危害仅为与产品密切相关的部分,并不能代表设备的全部风险。申请人应依据自身的质量管理体系要求,结合产品特点评估相关风险,并建立起科学全面的风险分析与管理制度,进而避免较大程度的风险及危害的发生。企业还应根据自身产品特点确定其他危害。针对产品的各项风险,企业应采取应对措施,确保风险降到可接受的程度。
2.呼吸湿化治疗相关设备在二类医疗器械产品注册申报中常见问题建议
由于企业对法规的理解深度不够,注册申报资料质量参差不齐,出现了很多共性问题,以下仅对该类产品注册过程中常见问题提出建议。
2.1 产品命名
产品仅满足GB 9706.1-2007标准要求时,不建议产品名称中含有“系统”一词,产品同时满足GB 9706.1-2007和GB 9706.15-2008标准时可在产品命名时使用“系统”一词。
2.2 产品类别
根据当前的技术水平,产品的技术原理不同时,应划分为不同的注册单元。例如空氧混合器,采用不同技术原理的,例如气动控制型空氧混合器与电动电控空氧混合器,应划分为不同的注册单元。技术原理相同,但产品设计结构不同的空氧混合器(例如不同的气路设计)建议划分为不同的注册单元。与呼吸湿化治疗仪、空氧混合器、医用呼吸道湿化器等配合使用的无源耗材(例如呼吸管路、气管插管、面罩等)建议与设备划分为不同的注册单元。
2.3 研究资料
2.3.1 生物学评价
与人体间接接触的部件(气体通路)如加热呼吸管路、加温湿化器、氧气连接管等和直接接触的部件如鼻氧管、血氧探头等,潜在生物学风险管理过程中的生物相容性评价工作,建议针对附件生物学评价情况分为以下三类:①豁免生物相容性评价的,建议参照国食药监械[2007]345号[3]要求,出具没有发生第四条第(一)款规定情况的说明性文件;②进行生物相容性评价的,建议按照GB/T 16886.1中系统方法框图所示的风险管理过程中生物学评价程序,对附件进行选择和评价;③进行生物相容性试验的,建议按照GB/T 16886.1附录,识别风险评定完整数据组需要补充的数据或试验。使用同类产品生物学数据的,企业需确认试验报告中的受试同类产品与申报产品在材料化学组成、各组成材料比例、产品物理结构、表面特性、生产工艺、灭菌方法、原材料供应商及技术规范、内包装材料(如适用)等任何可能影响生物相容性风险的因素均完全一致,并提供相关声明;若受试品与申报产品在以上所列可能影响生物相容性风险的因素中存在不一致的情况,则需提供充分的理由和证据支持所提交的试验报告适用于申报产品,必要时补充相应的生物学评价资料,如相关生物学试验项目的补充试验等;同类产品的生物学试验报告仅用于替代申报产品试验报告作为生物学评价的一部分,而不是替代申报产品的整体生物学评价报告。
2.3.2 稳定性研究
对于附件等耗材,提供货架有效期和包装研究资料,证明在货架有效期内,在生产企业规定的运输贮存条件下,产品可保持性能功能满足使用要求,具有微生物限度要求的产品还应当符合微生物限度要求,以无菌状态交付的产品还应保持无菌状态。
2.3.3 其他研究资料
呼吸湿化治疗类设备若具备脉搏氧饱和度测量功能,参照《脉搏氧饱和度测量仪注册技术审查指导原则》提交相应研究资料,研究验证应注意覆盖所有的适用人群。
2.4 临床评价
目前该类产品一般采用同品种评价途径,应关注同品种注册证的适用范围(作用部位的一致性)、不同工作模式的定义及用途、禁忌证,应针对产品每个功能模块、每种工作模式分别进行详细对比评价,包括工作原理、使用方法、适用人群、性能参数等,针对差异部分应分析差异不对产品安全有效性产生不利影响。提交同品种器械的临床经验数据应覆盖申报产品的工作模式(如CPAP、SMART等),关注多种工作模式的复合或协同治疗效果、研究结果的统计学意义,提交多份文献的可在文献中标注同品种产品的信息、用于临床研究的工作模式、应用人群、预期用途等位置。
2.5 产品技术要求及检验
第一,产品若具备CPAP等工作模式,应按照《正压通气治疗机注册技术审查指导原则》[4]的相关要求确认性能指标要求,该功能模式下的性能要求不应低于同品种产品的要求。
第二,流量、温度、氧浓度的监测性能建议量化指标,监测参数的性能指标应具体描述在哪个范围内能达到这样的误差水平。
第三,针对产品结构组成中的加温湿化器、加热呼吸管路、通气面罩、一次性血氧组件、氧气连接管和鼻氧管等部件:①若以非无菌状态供应的一次性使用部件应满足消毒级要求;②若以非无菌状态供应的重复性使用部件,除非在说明书中规定第一次使用前清洗消毒,否则也应满足消毒级要求:产品技术要求应规定微生物限度指标(包括初始污染菌、细菌菌落、致病菌等,参照GB 15979);③若以无菌状态供应的一次性使用部件,应在产品技术要求增加无菌及EO残留量(如适用)的要求。
3.小结
医疗器械风险管理是贯穿于医疗器械全生命周期的活动,对保证医疗器械的安全具有非常重要的作用。企业需要增强对风险管理的重视,技术审评人员需通过对风险点的认识,在注册申报中结合常见问题分析研究,从问题出发,结合医疗器械技术审评相关要求,提出建议,切实发挥风险管理为保障产品安全起到应有的作用,从而更好地把握产品的安全性、有效性、可靠性。
