野生二粒小麦主要农艺特性融入普通小麦的全基因组关联分析
2023-03-23龚方仪刘亚西颜泽洪钟晓英陈厚霖伍碧华
刘 佳 龚方仪 刘亚西 颜泽洪 钟晓英 陈厚霖 黄 林 伍碧华
野生二粒小麦主要农艺特性融入普通小麦的全基因组关联分析
刘 佳 龚方仪 刘亚西 颜泽洪 钟晓英 陈厚霖 黄 林*伍碧华*
西南作物基因资源发掘与利用国家重点实验室 / 四川农业大学小麦研究所, 四川成都 611130
野生二粒小麦(ssp.)是普通小麦的四倍体祖先种, 具有广泛的基因型变异, 是改良普通小麦的重要种质资源。本研究对不同年份和地点四个环境下的161份野生二粒小麦渗入系材料的株高、分蘖数、小穗数、抽穗期、开花期和千粒重进行表型鉴定, 并利用覆盖全基因组的13,116个DArT标记对各农艺性状进行全基因组关联分析, 以期发掘性状显著关联标记及相关候选基因。本研究共检测到147个与6个农艺性状相关的稳定标记。其中, 一些关联标记与抽穗期和开花期同时相关, 并且在2B染色体上聚集成簇。在野生二粒小麦渗入系群体中共推定了21个与农艺性状相关的候选基因, 其中位于7A染色体与千粒重显著相关的候选基因与细胞周期蛋白有关。这些标记和候选基因可为克隆优异农艺性状相关基因提供重要信息, 从而为野生二粒小麦在普通小麦背景中的遗传改良综合利用提供依据与指导。
野生二粒小麦; 普通小麦; 农艺性状; 远缘杂交; 全基因组关联分析
小麦是全球三大粮食作物之一, 世界上有1/3以上的人口以小麦为主粮, 特别是在发展中国家, 其产量和品质与人类的生存和健康息息相关[1]。随着经济和生活水平的快速提高以及人口的迅猛增长,全世界对小麦的需求量将持续增加, 高产育种一直是小麦现代育种永恒的主题[2-3]。
大量研究表明, 野生二粒小麦(, AABB, 2=4=28)是现代栽培小麦的祖先种, 起源“新月沃土”地区, 经历了长期复杂的环境演变形成其遗传多样性丰富的特点, 蕴藏着大穗、大籽粒、高粒重、多蘖性、理想的光合产量以及抗病、抗逆等许多优异的农艺与经济性状[4-6]。因此, 有学者认为野生二粒小麦是可被期望为全球小麦的可持续生产, 贡献其在数量和质量性状上全方位多样性的优异物种, 对小麦遗传改良具有重要的应用价值[5,7]。可惜的是, 野生二粒小麦丰富的遗传多样性在现代栽培小麦中已丢失近69%[8]。现已证明野生二粒小麦的A、B基因组与普通小麦同源, 易与普通小麦有性杂交进行遗传重组, 并且杂种后代与普通小麦容易回交, 从而使得优异基因可以通过同源重组而成功导入普通小麦, 进而能够有效丰富小麦的遗传变异, 极大拓展了普通小麦的遗传基础[9]。储诚艮等[10]研究表明, 通过野生二粒小麦与普通小麦的杂交、回交等可将野生二粒小麦的大粒和籽粒高蛋白质含量(GPC)成功向普通小麦转移, 并实现两者的同步改良。我们前期研究发现绝大部分含野生二粒小麦血缘的渐渗系不仅GPC 显著高于其普通小麦受体品种川农16 (CN16), 一些甚至达到了优质强筋或中强筋小麦品种的GPC水平, 而且其籽粒更大、产量更高[11-14]。在此基础上, 挖掘出一些除基因之外的高GPC位点和差异表达基因, 分析并验证了一些候选基因的功能[14-16]。然而, 迄今对于野生二粒小麦优异大粒特性显著增大普通小麦的籽粒库容和粒重, 获得高产[17]的优异位点及其候选基因尚知之甚少。
因此, 本研究在结合我们前期工作的基础上, 利用野生二粒小麦为父本与普通小麦川农16远缘杂交创建的稳定高世代野生二粒小麦渗入系群体, 结合分子标记技术对主要农艺性状进行全基因组关联分析, 挖掘与农艺性状显著关联的标记位点; 探讨野生二粒小麦农艺性状在普通小麦背景下的遗传与重组情况, 以及野生二粒小麦优异遗传物质向普通小麦的导入能力, 及其在普通小麦背景中的进一步传递与表达能力, 为野生二粒小麦在普通小麦背景中的遗传改良综合利用提供依据与指导。
1 材料与方法
1.1 试验材料
本研究的试验材料为引自以色列的含有高籽粒蛋白含量的野生二粒小麦D1作为父本与普通小麦矮杆、穗数型(千粒重小)、弱筋国审品种川农16 (CN16) (https://xms.sicau.edu.cn/info/1071/2580.htm)远缘杂交创建的稳定高世代野生二粒小麦渗入系群体。所有供试材料均保存在四川农业大学小麦研究所。161份D1渗入系群体来源包括: (1) CN16与野生二粒小麦D1杂交得到的具有稳定42条染色体的106个高代株系(F12) (子一代)。(2) 随机选取子一代再分别与不同的普通小麦品种绵麦46 (MM46)、川麦50 (CM50)、科成麦2号(KCM2)和川育18 (CY18)/云B58863 (YB58863)杂交后获得的55个高代株系(F10)(子二代) (附表1)。
1.2 试验设计
供试材料于2014年10月和2015年10月种植于四川农业大学温江农场和崇州基地。采用完全随机区组设计, 单粒播种, 行长2 m, 行间距30 cm, 株间距10 cm, 每个点设置3次重复, 边行设置保护行。田间管理与大田生产基本一致。2014—2015年温江和崇州点数据分别用2015温江和2015崇州表示, 2015—2016年温江和崇州点数据分别用2016温江和2016崇州表示。
1.3 农艺性状调查与测定
成熟时每行材料排除边际效应后从中间随机选取6株, 调查测定其株高、分蘖数、小穗数和千粒重。株高以主茎高表示即自地面至穗顶(不含芒)的高度, 取其平均值, 单位为cm。有效分蘖和小穗数以蜡熟期植株的第1个茎和穗上为节点进行测定。抽穗期以每份材料50%的植株穗由旗叶叶鞘露出二分之一, 记录当天日期。开花期以每份材料50%的植株开花后记录当天日期。千粒重测定参照GB/T 5519- 2008标准, 收获脱粒后用电子天平进行测定, 3次生物学重复[18]。使用SPSS 19.0软件(IBM, 美国)用于各农艺性状的相关性分析和方差分析。
1.4 DArT标记基因分型
将基因组DNA样品提供给澳大利亚Diversity Arrays Technology Pty Ltd.公司(澳大利亚, http:// www.Triticarte.com.au/)选择小麦上开发的DArT标记进行扫描分型。获得的原始数据基于调用率(最小阈值85%)和重现性(最小阈值95%)进行质量控制, 其次将缺失率大于10%和次要等位基因频率(MAF)小于5%的标记过滤, 最终获得用于后续分析的DArT标记[19]。
1.5 群体结构和连锁不平衡(LD)分析
使用基于贝叶斯聚类模型为运算基础的Structure v2.3.4软件计算相应材料的Q值(第材料的基因组变异源于第群体的概率)来评估群体结构[20]。采用admixed model, 亚群数K值范围设置为1~10, 进行5次迭代, 运行时将MCMC (Markov Chain Monte Carlo)的不作数迭代(Length of Bum-in Period)设为100,000次。同时, 使用TASSEL 3.0进行过滤标记的主成分分析(PCA)和LD分析[21]。
1.6 全基因组关联分析(GWAS)及相关候选基因预测
为消除环境影响, 使用META-R进行了所有测试环境下的最佳线性无偏预测(BLUP)[22]。利用TASSEL中的一般线性模型(GLM)和混合线性模型(MLM)进行DArT标记和性状的关联分析, 设置值的–log10()≥3.00的阈值为性状标记显著关联[23]。使用Haploview4.2软件作曼哈顿图显示显著MTA, quantile-quantile图展示重要的值分布[24]。利用国际小麦基因组测序联盟数据库(IWGSC; http://www. wheatgenome.org/)、国际野二粒小麦基因组测序联盟数据库(http://wewseq.wixsite.com/consortium)和小麦族多组学中心数据库(http://202.194.139.32/)进行BLAST比对鉴定相关候选基因。
2 结果与分析
2.1 表型分析
野生二粒小麦渗入系群体及其亲本CN16和D1的农艺性状测定结果详见表1。所有测试环境中, 双亲的株高差异较显著, 其中野生二粒小麦D1的株高平均范围为92.20~120.37 cm, 高于CN16 (79.73~ 87.60 cm) (表1)。野生二粒小麦D1渗入系群体株高变化范围较大, 其中最低的是2015崇州的材料为70.40 cm, 最高的是2015温江的材料为123.30 cm, 表现出丰富的遗传多样性。野生二粒小麦D1渗入系群体在2015温江、2015崇州、2016温江和2016崇州4个环境中的株高平均值分别为99.03、97.35、94.42和90.88 cm, 表现为中秆(90~100 cm)水平。方差分析表明, 野生二粒小麦D1渗入系群体在2015崇州中株高均值显著高于亲本CN16, 而在其余3个环境中的株高均值与亲本CN16的差异性不显著(表1)。
所有测试环境中, 双亲的分蘖数的差异较显著, 其中野生二粒小麦D1的分蘖数均值范围为9.53~ 24.13个, 显著高于CN16 (7.60~10.67个) (表1)。野生二粒小麦D1渗入系群体的分蘖数在4个环境中的均值范围为7.51~9.54个。方差分析表明, 野生二粒小麦D1渗入系群体在所有环境中的均值分蘖数达到亲本CN16的水平。其中, 分蘖数最低的是2016温江的材料为4.30个, 最高的是2016崇州的材料为15.90个(表1)。
所有测试环境中, 野生二粒小麦D1的小穗数均值范围为19.27~20.97个, CN16的小穗数均值范围为17.07~21.13个, 在2015年2个环境下野生二粒小麦D1的小穗数均值显著高于CN16, 但是在2016年2个环境下二者的差异性不显著(表1)。野生二粒小麦D1渗入系群体小穗数均值范围为19.33~19.98个, 其中小穗数最少的材料为15.90个出现在2016温江环境中, 最多的材料为24.10个出现在2015温江环境中。方差分析结果表明, 野生二粒小麦D1渗入系群体在所有环境中的小穗数均值与亲本野生二粒小麦D1的差异不显著, 除3个环境中的小穗数均值与亲本CN16的相当外, 2015崇州的还显著高于CN16 (表1)。
所有环境中, 双亲的抽穗期差异显著, 其中野生二粒小麦D1的抽穗期均值范围为152.00~165.00 d, 显著长于CN16 (124.00~142.67 d) (表1)。野生二粒小麦D1渗入系群体的抽穗期变化范围较大, 其中抽穗期天数最短的是2015崇州的材料为121.30 d, 最长的是2016温江的材料为152.30 d, 表现出丰富的遗传多样性。野生二粒小麦D1渗入系群体在4个环境中的抽穗期均值范围为128.60~145.18 d。方差分析结果表明, 野生二粒小麦D1渗入系群体在所有环境中的抽穗期均值显著短于野生二粒小麦D1, 而与亲本CN16的相当(表1)。
所有环境中, 双亲的开花期差异显著, 其中野生二粒小麦D1的开花期均值范围为158.00~170.33 d, 显著长于CN16 (136.67~154.67 d) (表1)。野生二粒小麦D1渗入系群体的开花期变化范围较大, 其中开花期天数最短的是2015崇州的材料为133 d, 最长的是2016温江的材料为161.70 d, 表现出丰富的遗传多样性。野生二粒小麦D1渗入系群体在4个环境中的开花期均值范围为140.31~156.67 d。方差分析结果表明, 野生二粒小麦D1渗入系群体在所有环境中的开花期均值显著短于野生二粒小麦D1, 而与亲本CN16相当(2015温江环境除外) (表1)。
双亲的千粒重在所有环境中的差异性显著, 其中亲本CN16的千粒重均值范围为42.70~49.33 g, 显著高于野生二粒小麦D1 (15.00~32.80 g) (表1)。野生二粒小麦D1渗入系群体在4个环境中的千粒重均值范围为46.08~52.90 g。野生二粒小麦D1渗入系群体的千粒重变异范围较大, 其中千粒重最低的是2015温江的材料为30.70 g, 最高的是2015崇州的材料为64.63 g, 表现出丰富的遗传多样性。方差分析结果表明, 野生二粒小麦D1渗入系群体在所有环境中的千粒重均值显著高于野生二粒小麦D1, 同时在2016崇州环境下显著高于亲本CN16, 而在其余3个环境中与亲本CN16的水平相当(表1)。
本研究中野生二粒小麦渗入系群体的6个主要农艺性状频率分布结果显示, 在所有环境中株高的最高频率分布区域主要集中在90~110 cm, 表现为中秆(90~100 cm)和中高秆(100~110 cm)水平; 分蘖数的最高频率分布区域是6~10个; 小穗数的最高频率分布区域在19~20个; 千粒重的最高频率分布区域主要在45~55 g范围内。然而抽穗期和开花期的最高频率分布区域在各个环境中都不相同, 但二者在各环境中的分布规律一致。其中, 抽穗期和开花期在2015温江和2016崇州环境中的最高频率分布区域分别是135~140 d和143~148 d, 在2015崇州环境中的最高频率分布区域分别是125~130 d和138~143 d, 在2016温江环境中的最高频率分布区域分别是140~145 d和153~158 d (图1)。

表1 野生二粒小麦D1渗入系群体及其双亲农艺性状的表型变异
小写字母a、b和c表示相同环境的渗入系群体及其双亲性状间在0.05概率水平差异显著。
Lowercase letters a, b, and c indicate significant difference at the 0.05 probability level between introgression lines and its parents trait in the same environment.
基于BLUP数据的相关性分析结果显示, 野生二粒小麦D1渗入系群体的株高分别与分蘖数和千粒重呈显著正相关, 其相关系数分别为0.162 (<0.05)和0.300 (<0.01), 而与小穗数呈极显著负相关, 相关系数为–0.351 (<0.01); 小穗数与分蘖数呈极显著负相关, 相关系数为–0.342 (<0.01); 小穗数与抽穗期和开花期呈极显著正相关, 相关系数分别为0.311和0.209 (<0.01), 而与千粒重呈显著负相关, 相关系数为–0.169 (<0.05); 抽穗期和开花期呈极显著正相关, 并且二者相关系数最高,=0.938 (<0.01) (表2)。
2.2 群体结构和连锁不平衡分析
基于群体结构和连锁不平衡LD分析, 野生二粒小麦渗入系群体可分为3个主要亚群; A、B、D基因组以及整体的平均2值随着遗传距离的增加而迅速衰减, 对于A、B、D亚基因组以及全基因组的LD衰减距离分别约为9、12、13和12 cM, 与前期研究结果描述一致[14]。
2.3 GWAS分析
GLM模型下共检测到5176个MTA分布在21条染色体上, 与株高、分蘖数、小穗数、抽穗期、开花期和千粒重相关的显著性状关联标记(MTA)分别有1162、932、1566、529、630和357个, 其中与分蘖数相关的MTA的表型变异解释率(PVE)最高为21.60% (表3)。MLM模型下共检测到349个MTA, 与株高、分蘖数、小穗数、抽穗期、开花期和千粒重相关MTA分别有25、59、6、188、26和45个(表3)。其中, 在两个模型中均检测到147个稳定的MTA,具体结果如下:
两个模型一致检测到14个株高的稳定MTA, 它们位于1A、1B、2A、2B、3A、6D、7A和7B染色体上, 其中位于3A和6D染色体的MTA 在两模型中的PVE值相对较高; 与分蘖数相关的55个稳定MTA,分别定位在1B、2B、2D、3B、4A、4B、4D、6A、6B、7A和7D染色体上, 其中位于4B染色体的MTA在GLM和MLM中的PVE值最高, 分别为21.60%和8.04%; 与小穗数相关的5个稳定MTA, 分别定位在3A、4A、7B和7D染色体上, 其中位于7D染色体的MTA1111098在GLM和MLM中的PVE值最高, 分别为21.18%和8.81%; 与抽穗期相关的39个稳定MTA, 分别定位于1A、2B、2D、4D、5D、6A、6B、7A和7B染色体上, 其中在2B上检测到的MTA最多;与开花期相关的19个稳定MTA, 分别定位于2B、2D、3B、4B和7D染色体上, 其中在2B上检测到的MTA最多, 并且2B染色体上的13个MTA与抽穗期中检测到的相同; 与千粒重相关的15个稳定MTA, 分别定位于3B、5A、6A、7A和7D染色体上, 其中在3B上检测到的MTA最多(附表2)。此外, 从GWAS分析获得的曼哈顿和Quantile-Quantile图(图2和图3)分析可见, 与GLM的结果相比, MLM的表现更加保守。
为进一步明确亲本野生二粒小麦在渗入系群体中的贡献情况, 我们对已经获得的稳定MTA进行筛选分析, 结果表明在这些关联标记中, 源自野生二粒小麦A、B基因组的标记共有45个, 其中2B染色体上最多(27个), 主要影响分蘖数、开花期和抽穗期。其中与株高相关的6个稳定MTA, 分别定位在1A、1B、2A、3A和7B染色体上; 位于5A和7A染色体上的2个稳定MTA主要影响千粒重(表4)。

表2 野生二粒小麦D1渗入系群体的农艺性状基于BLUP数据的皮尔森相关性分析
*相关性在0.05概率水平差异显著,**相关性在0.01概率水平差异显著。
*correlation is significant at the 0.05 probability level;**correlation is significant at the 0.01 probability level.

图1 野生二粒小麦D1渗入系群体的农艺性状在各环境下的频率分布
实线箭头代表亲本野生二粒小麦D1, 虚线箭头代表亲本普通小麦CN16。WJ: 温江; CZ: 崇州。
The solid arrow indicates the parent wild emmer wheat D1, and the dotted arrow indicates the parent common wheat CN16. WJ: Wenjiang; CZ: Chongzhou.

表3 野生二粒小麦D1渗入系群体在GLM和MLM模型下的农艺性状全基因组关联分析
a: GLM与MLM两种模型分别检测到的显著MTA个数;b: GLM与MLM两种模型都检测到的显著MTA个数; MLM: 混合线性模型, GLM: 一般线性模型。
a: the total number of MTAs identified by GLM and MLM, respectively;b: the number of significant and stable MTAs detected by GLM and MLM; MLM: the mixed linear model; GLM: the general linear model.

图2 基于混合线性模型下野生二粒小麦D1渗入系群体农艺性状的全基因组关联曼哈顿扫描图
A: 混合线性模型检测到的显著MTA, 红线表示–log10()阈值为3.00; B: Quantile-Quantile图中黑线表示预期值。
A: chromosomes carrying significant MTA detected by MLM, the red line indicates the –log10() threshold of 3.00; B: the black line indicates the expected values in Quantile-Quantile plots.
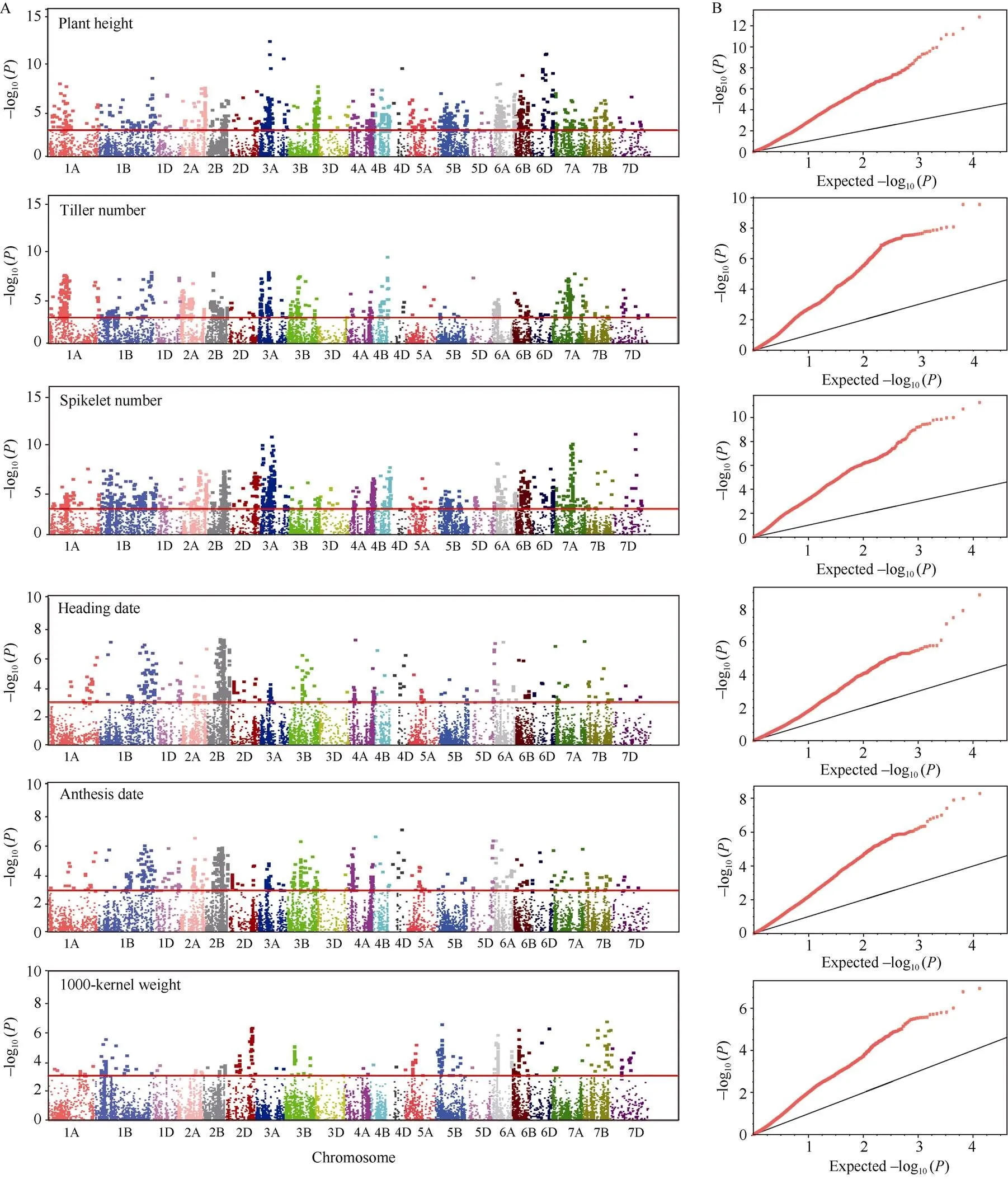
图3 基于一般线性模型下野生二粒小麦D1渗入系群体农艺性状的全基因组关联曼哈顿扫描图
A: 一般线性模型检测到的显著MTA, 红线表示–log10()阈值为3.00; B: Quantile-Quantile图中黑线表示预期值。
A: chromosomes carrying significant MTA detected by GLM, the red lines indicate the –log10() threshold of 3.00; B: the black lines indicate the expected values in Quantile-Quantile plots.

表4 源于野生二粒小麦A、B基因组的性状显著关联标记
MTA: marker-trait associations.
2.4 相关候选基因预测
使用中国春小麦基因组参考序列(RefSeq v.1.0)和野生二粒小麦基因组参考序列(WEWSeq v.1.0)对表现稳定的MTA进行比对, 共推定了21个与6个农艺性状相关的候选基因。株高、分蘖数、小穗数、抽穗期、开花期和千粒重相关的候选基因分别有5、4、3、7、7和1个(附表3)。株高相关的5个基因由生长素响应因子、钙敏感受体和果胶裂解酶家族蛋白构成; 分蘖数相关的基因包括热激蛋白DnaJ、富含甘氨酸的细胞壁结构蛋白和磷酸葡萄糖/磷酸盐转运子; 与小穗数相关的基因有氨基酸转运蛋白和多核苷酸转移酶; 与抽穗期相关的基因由位于2B染色体的糖转运蛋白、钙依赖性蛋白激酶(CDPKs)、驱动蛋白、富含亮氨酸的蛋白激酶家族蛋白、F-box蛋白、抗病蛋白和位于4D染色体的乙烯反应转录因子构成, 其中位于2B染色体的6个基因同时也与开花期性状相关; 此外与开花期相关的基因还包括MADS-box转录因子; 千粒重相关的候选基因与细胞周期蛋白有关(附表3)。
3 讨论
3.1 野生二粒小麦主要农艺性状在普通小麦背景下的响应
在我们前期研究工作的基础上, 本研究通过所创制的野生二粒小麦渗入系群体从分子水平进一步揭示野生二粒小麦的农艺性状(千粒重、株高、分蘖数、小穗数、抽穗期和开花期)在普通小麦背景下的遗传和重组情况。
总体上, 野生二粒小麦渗入系群体的株高和亲本野生二粒小麦D1相比已经显著降低, 大多数材料都处于中秆和中高秆水平, 并且与分蘖数和千粒重呈显著正相关, 这和前人的研究结论相一致[25-26]。有关研究表明, 小麦株高与植株抗倒伏能力、产量和品质之间都有一定相关性, 适宜的株高有利于形成较多的分蘖数、增加千粒重, 从而有效提高小麦产量[27-29]。此外, 野生二粒小麦渗入系群体的抽穗期和开花期与亲本CN16的差异不显著, 但是显著短于野生二粒小麦D1, 同时分蘖数和小穗数基本达到了亲本普通小麦CN16的水平。这些结果都表明野生二粒小麦渗入系群体的株高、分蘖数、小穗数、开花期和抽穗期性状都在向着现代小麦育种目标有利的方向发展。特别是千粒重也同样达到了亲本普通小麦CN16的水平, 甚至大部分材料的千粒重都超过了双亲本身。小麦产量构成的三因素中, 千粒重对最终产量的影响起着主要作用, 而小麦粒重的高低主要决定于籽粒体积(即库容)的大小[30]。本研究中的亲本普通小麦CN16是矮秆、穗数型(千粒重小)的国审高产品种, 这暗示野生二粒小麦渗入系群体千粒重增加的主要缘由在于其籽粒库容的增大, 因为野生二粒小麦籽粒长度明显长于普通小麦CN16, 而CN16的籽粒宽度和厚度大于野生二粒小麦, 经过它们远缘杂交产生的渗入系后代由于整合了双亲籽粒的优良特性, 引起籽粒体积在三维空间上进一步扩大, 使得籽粒库容得以改良, 进而增加籽粒千粒重而提高产量, 这充分证实了我们之前对于野生二粒小麦能够改良普通小麦库容性的推测[17]。显然, 野生二粒小麦基因的导入, 能使普通小麦栽培品种的千粒重得到很好改良, 对提高小麦产量具有很大潜力, 而且所创制渗入系的综合农艺性状也符合高产育种目标, 这与我们之前关于野生二粒小麦在改良普通小麦的品质、抗性的同时, 不但没有伤及反而提高了高产相关农艺性状的结果具有一致性[11-13,15-17]。
3.2 农艺性状显著关联位点及候选基因
对于农艺性状相关位点的研究前人已经有很多报道, 相关学者在不同的群体中检测到控制株高的QTL分布在1B、2D、3B、5A、5B、6A和7D染色体上[31-32]。我们在1B染色体上同样检测到株高关联的MTA。Sourdille等[33]在2B和7B上发现了控制抽穗期的位点, Huang等[34]在2A、2D、3B、5A、6A和7B上定位了7个抽穗期相关QTL, 本研究在2B、2D、6A和7B上也检测到了相似的位点。Börner等[35]在2B、2D、5A和5D上定位了控制开花期的位点, 本研究也在2B、2D上检测到开花期关联的MTA。此外, 本研究在野生二粒小麦渗入系群体的2B染色体上检测到与抽穗期和开花期同时相关的MTA区段, 而且发现控制抽穗期和开花期的MTA在染色体上集聚成簇, 推测可能是由于单基因多效或者是多基因紧密连锁共同控制这两个性状, 至于其具体缘由尚需进一步探究。本研究中在3B、5A、6A、7A和7D染色体上发现的千粒重相关MTA, 在前人报道的相应染色体上也均有发现[36-38]。
本研究在供试的野生二粒小麦渗入系群体中推定得到21个性状相关的候选基因。其中, 与株高相关的基因与生长素响应因子和钙敏感受体等相关。有研究表明生长素响应因子与植物的生长发育密切相关, 如植物的向性运动、顶端优势、侧根发生、细胞伸长、细胞分裂及细胞分化等都有生长素响应因子的参与[39]。本研究发现分蘖数相关的基因包括热激蛋白DnaJ和富含甘氨酸的细胞壁结构蛋白等。李春喜和赵广才[40]报道富含甘氨酸的一类超级蛋白家族在植物中广泛存在并与激素响应相关, 而小麦分蘖的变化动态又与内源激素关系密切。黄祥富等[41]研究发现热激蛋白与植物种子发育也密切相关。因此我们推测热激蛋白在一定程度上也影响了野生二粒小麦渗入系的分蘖和小穗数。野生二粒小麦渗入系群体中位于2B染色体的抽穗期和开花期相关基因与F-box蛋白和钙依赖性蛋白激酶(CDPKs)等有关, 而已有研究表明, F-box基因家族是植物中最大的基因家族之一, 其基因编码的蛋白能够调节多种多样的生命活动, 如延缓植物衰老、调控植物开花等[42]。同时, 我们检测到与开花期相关的基因中还包括MADS-box转录因子, 现已证明MADS- box基因是禾本科植物(如大麦、小麦等)开花调控网络中最重要的转录调控因子[43]。本研究结果还表明, 与千粒重相关的基因和细胞周期蛋白有关, 而前人研究发现细胞周期蛋白在细胞周期调控中具有新功能, 并指出通过调节细胞周期蛋白能够提高谷物的产量[44]。在未来的研究中需要进行功能分析以进一步了解这些候选基因调控小麦相关农艺性状的途径。本研究中鉴定的农艺性状显著MTA以及相关的候选基因可为克隆优异农艺性状相关基因提供重要信息, 用于分子标记辅助育种, 从而为综合利用野生二粒小麦进行普通小麦遗传改良提供重要的理论依据。
4 结论
从野生二粒小麦渗入系群体中共检测到147个与6个农艺性状相关的稳定标记, 并推定了21个候选基因。其中, 与抽穗期和开花期同时相关的标记在2B染色体上聚集成簇, 位于7A染色体与千粒重显著相关的候选基因与细胞周期蛋白有关, 该基因可能参与调控千粒重。
附表 请见网络版: 1) 本刊网站http://zwxb. chinacrops.org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical- zuowxb.aspx。
[1] 赵广才, 常旭虹, 王德梅, 陶志强, 王艳杰, 杨玉双, 朱英杰. 小麦生产概况及其发展. 作物杂志, 2018, 34(4): 1–7.
Zhao G C, Chang X H, Wang D M, Tao Z Q, Wang Y J, Yang Y S, Zhu Y J. General situation and development of wheat production., 2018, 34(4): 1–7 (in Chinese with English abstract).
[2] International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat () genome., 2014, 345: 1251788.
[3] Jones J M, Pena R J, Korczak R, Braun H J. Carbohydrates, grains, and wheat in nutrition and health: an overview Part II. Grain terminology and nutritional contributions., 2015, 60: 260–271.
[4] Joppa L R, Cantrell R G. Chromosomal location of genes for grain protein content of wild tetraploid wheat., 1990, 30: 1059–1064.
[5] Jaradat A A. Ecogeography, genetic diversity, and breeding value of wild emmer wheat (‘’ korn ex Asch. and Graebn.) Thell., 2011, 5: 1072.
[6] Van Silfhout C H, Gerechter-Amitai Z K. Adult-plant resistance to yellow rust in wild emmer wheat., 1988, 94: 267–272.
[7] Nevo E. Genetic resources of wild emmer,, for wheat improvement in the third millennium., 2001, 49: 77–92.
[8] Haudry A, Cenci A, Ravel C, Bataillon T, Brunel D, Poncet C, Hochu I, Poirier S, Santoni S, Glémin S, David J. Grinding up wheat: a massive loss of nucleotide diversity since domestication., 2007, 24: 1506–1517.
[9] Kushnir U, Halloran G M. Transfer of high kernel weight and high protein from wild tetraploid wheat () to bread wheat () using homologous and homoeologous recombination., 1984, 33: 249–255.
[10] 储诚艮, 冯祎高, 陈佩度. 将野生二粒小麦的大粒和籽粒高蛋白含量性状向普通小麦的转移. 南京农业大学学报, 2001, 24(2): 16–19.
Chu C G, Feng Y G, Chen P D. The transference of the traits large kernel and high seed protein content frominto common wheat., 2001, 24: 16–19 (in Chinese with English abstract).
[11] 胡喜贵. 关切小麦加工和营养品质的和位点的种质资源鉴定及利用研究. 四川农业大学博士学位论文, 四川成都, 2013.
Hu X G. Characterization and Utilization ofandLoci Associated with Wheat Processing and Nutritional Quality inand Its Related Species. PhD Dissertation of Sichuan Agricultural University, Chengdu, Sichuan, China, 2013 (in Chinese with English abstract).
[12] Jiang Z L, Wu B H, Wang Z Z, Hu J L, Yuan J, Chen H L, Liu J, Zheng Y L, Liu D C. Enriching novelalleles and significantly strengthening gluten properties of common wheat through wide hybridization with wild emmer., 2017, 76: 271–279.
[13] Wang Z Z, Huang L, Wu B H, Hu J L, Jiang Z L, Qi P F, Zheng Y L, Liu D C. Characterization of an integrated activeallele in common wheat from wild emmer and its potential role in flour improvement., 2018, 19: 923.
[14] Liu J, Huang L, Wang C Q, Liu Y X, Yan Z H, Wang Z Z, Xiang L, Zhong X Y, Gong F Y, Zheng Y L, Liu D C, Wu B H. Genome-wide association study reveals novel genomic regions associated with high grain protein content in wheat lines derived from wild emmer wheat., 2019, 10: 464–464.
[15] Gong F Y, Qi T G, Zhang T, Lu Y S, Liu J, Zhong X Y, He J S, Li Y F, Zheng Y L, Liu D C, Huang L, Wu B H. Comparison of the agronomic, cytological, grain protein characteristics, as well as transcriptomic profile of two wheat lines derived from wild emmer., 2022, 12: 804481.
[16] Gong F Y, Huang L, Qi T G, Tang G, Liu J, Xiang L, He J S, Zheng Y L, Liu D C, Wu B H. Comparative analysis of developing grain transcriptome reveals candidate genes and pathways improving GPC in wheat lines derived from wild emmer., 2021, 62: 17–25.
[17] 伍碧华, 路洁霏, 李平, 胡喜贵, 郑有良. 野生二粒小麦籽粒蛋白质含量和库容性状的遗传多样性与可利用性研究. 四川农业大学学报, 2008, 26: 221–225.
Wu B H, Lu J F, Li P, Hu X G, Zheng Y L. Genetic diversity and potential utilization of grain protein content and sink traits in., 2008, 26: 221–225 (in Chinese with English abstract).
[18] 国家粮食局. 谷物与豆类千粒重的测定, GB T 5519-2008, 2008.
State Administration of Grain. Cereals and Pulses-Determination of the Mass of 1000-grain, GB T 5519-2008, 2008 (in Chinese).
[19] Liu Y X, Lin Y, Gao S, Li Z Y, Ma J, Deng M, Chen G Y, Wei Y M, Zheng Y L. A genome-wide association study of 23 agronomic traits in Chinese wheat landraces., 2017, 91: 861–873.
[20] Pritchard J K, Stephens M, Rosenberg N A, Donnelly P. Association mapping in structured populations., 2000, 67: 170–181.
[21] Bradbury P J, Zhang Z, Kroon D E, Casstevens T M, Ramdoss Y, Buckler E S. TASSEL: software for association mapping of complex traits in diverse samples., 2007, 23: 2633–2635.
[22] Alvarado G, López M, Vargas M, Pacheco Á, Rodríguez F, Burgueño J, Crossa J. META-R (Multi Environment Trail Analysis with R for Windows) Version 6.04. Mexico, International Maize and Wheat Improvement Center, 2015 [2022-02-17]. https:// data.cimmyt.org/dataset.xhtml? persistentId=hdl:11529/10201.
[23] Alomari D Z, Eggert K, Von Wirén N, Alqudah A M, Polley A, Plieske J, Ganal M W, Pillen K, Röder M S. Identifying candidate genes for enhancing grain Zn concentration in wheat., 2018, 9: 1313.
[24] Barrett J C, Fry B, Maller J, Daly M J. Haploview: analysis and visualization of LD and haplotype maps., 2004, 21: 263–265.
[25] 段国辉, 高海涛, 张学品, 吴少辉, 杨洪强, 王艳芳. 冬小麦水旱条件下株高构成与产量性状及抗旱指数相关分析. 陕西农业科学, 2006, (4): 1–3.
Duan G H, Gao H T, Zhang X P, Wu S H, Yang H Q, Wang Y F. Correlation analysis of plant height composition, yield characters and drought resistance index of winter wheat under flood and drought conditions., 2006, (4): 1–3 (in Chinese).
[26] 贾继增. 小麦粒重与植株性状相关因素的统计分析. 作物学报, 1984, 10: 201–205.
Jia J Z. The statistical analysis for correlation factors in kernel weight and plant characters in wheat (L.)., 1984, 10: 201–205 (in Chinese with English abstract).
[27] Austin R B, Bingham J, Blackwell R D, Evans L T, Ford M A, Morgan C L, Taylor M. Genetic improvements in winter wheat yields since 1900 and associated physiological changes., 1980, 94: 675–689.
[28] 高士杰. 作物株型改良的增产效用. 吉林农业科学, 1999, 24(2): 23–24.
Gao S J. Yield-increasing effect of crop plant type improvement., 1999, 24(2): 23–24 (in Chinese).
[29] 盛承师. 小麦冠层形态结构与籽粒产量的关系(三): 理想株型的设计. 国外农学: 麦类作物, 1987, (1): 35–38.
Sheng C S. Relationship between wheat canopy morphology and grain yield: 3. Design for ideal plant type.:, 1987, (1): 35–38 (in Chinese).
[30] 张智猛. 华北地区小麦生育进程、灌浆特性与库容对粒重相对重要性的研究. 河北农业大学学报, 1998, (2): 22–27.
Zhang Z M. Studies on the contribution of growing process, filling properties, storage capacity to grain weight for wheat in North China., 1998, (2): 22–27 (in Chinese with English abstract).
[31] 李俊周, 刘艳阳, 何宁, 崔党群. 小麦DH群体数量性状的遗传分析. 麦类作物学报, 2005, 25(3): 16–19.
Li J Z, Liu Y Y, He N, Cui D Q. Genetics analysis of several quantitative traits of doubled haploid population in wheat., 2005, 25(3): 16–19 (in Chinese with English abstract).
[32] 李战怡. 西南小麦地方品种农艺性状评价及关联分析. 四川农业大学硕士学位论文, 四川成都, 2015.
Li Z Y. Agronomic Traits Evaluation and Association Analysis of Wheat Landraces in Southwest China. MS Thesis of Sichuan Agricultural University, Chengdu, Sichuan, China, 2015 (in Chinese with English abstract).
[33] Sourdille P, Snape J W, Cadalen T, Charmet G, Nakata N, Bernard S, Bernard M. Detection of QTLs for heading time and photoperiod response in wheat using a doubled-haploid population., 2000, 43: 487–494.
[34] Huang X Q, Cloutier S, Lycar L, Radovanovic N, Humphreys D G, Noll J S, Somers D J, Brown P D. Molecular detection of QTLs for agronomic and quality traits in a doubled haploid population derived from two Canadian wheats (L.)., 2006, 113: 753–766.
[35] Börner A, Schumann E, Fürste A, Cöster H, Leithold B, Röder M, Weber W. Mapping of quantitative trait loci determining agronomic important characters in hexaploid wheat (L.)., 2002, 105: 921–936.
[36] Campbell K G, Bergman C J, Gualberto D G, Anderson J A, Giroux M J, Hareland G, Fulcher R G, Sorrells M E, Finney P L. Quantitative trait loci associated with kernel traits in a soft × hard wheat cross., 1999, 39: 1184–1195.
[37] Ammiraju J S S, Dholakia B B, Santra D K, Singh H, Lagu M D, Tamhankar S A, Dhaliwal H S, Rao V S, Gupta V S, Ranjekar P K. Identification of inter simple sequence repeat (ISSR) markers associated with seed size in wheat., 2001, 102: 726–732.
[38] 郑有良, 颜济杨俊良. 小麦粒重基因定位研究. 作物学报, 1993, 19: 304–308.
Zheng Y L, Yan J, Yang J L. Localization of genes for grain weight in common wheat., 1993, 19: 304–308 (in Chinese).
[39] 刘振华, 于延冲, 向凤宁. 生长素响应因子与植物的生长发育.遗传, 2011, 33: 1335–1346.
Liu Z H, Yu Y C, Xiang F N. Auxin response factors and plant growth and development.(Beijing),2011, 33: 1335–1346 (in Chinese with English abstract).
[40] 李春喜, 赵广才. 小麦分蘖变化动态与内源激素关系的研究. 作物学报, 2000, 26: 963–968.
Li C X, Zhao G C. Research on the relationship between wheat tillering dynamics and endogenous hormone., 2000, 26: 963–968 (in Chinese with English abstract).
[41] 黄祥富, 黄上志, 傅家瑞. 植物热激蛋白的功能及其基因表达的调控. 植物学报, 1999, 16: 530–536.
Huang X F, Huang S Z, Fu J R. Regulation of expression and functions of the heat shock proteins of plant., 1999, 16: 530–536 (in Chinese with English abstract).
[42] 许克恒, 张云彤, 张莹, 王彬, 王法微, 李海燕. 植物F-box基因家族的研究进展. 生物技术通报, 2018, 34(1): 26–32.
Xu K H, Zhang Y T, Zhang Y, Wang B, Wang F W, Li H Y. Research advances on the f-box gene family in plants., 2018, 34(1): 26–32 (in Chinese with English abstract).
[43] 李元元, 王鲁, 苏振刚, 王元英. MADS-box基因控制植物成花的分子机理. 基因组学与应用生物学, 2010, 29: 1122–1132.
Li Y Y, Wang L, Su Z G, Wang Y Y. The molecular mechanism of MADS-box genes regulates floral formation and flowering in plant., 2010, 29: 1122–1132 (in Chinese with English abstract).
[44] Qi P, Lin Y S, Song X J, Shen J B, Huang W, Shan J X, Zhu M Z, Jiang L, Gao J P, Lin H X. The novel quantitative trait locuscontrols rice grain size and yield by regulating Cyclin-T1;3., 2012, 22: 1666.
Genome-wide association study for agronomic traits in common wheat lines derived from wild emmer wheat
LIU Jia, GONG Fang-Yi, LIU Ya-Xi, YAN Ze-Hong, ZHONG Xiao-Ying, CHEN Hou-Lin, HUANG Lin*,and WU Bi-Hua*
State Key Laboratory of Crop Gene Exploration and Utilization in Southwest China / Triticeae Research Institute, Sichuan Agricultural University, Chengdu 611130, Sichuan, China
Wild emmer (ssp.) is the tetraploid progenitor of common wheat with wide genotypic variations, which is an important germplasm resource for common wheat improvement. In this study, the plant height, tiller number, spikelet number, heading date, anthesis date, and 1000-kernel weight of 161introgression lines derived from wild emmer were phenotyped in four environments of different years and locations. We performed genome-wide association study (GWAS) of agronomic traits using 13,116 DArT markers to identify marker-trait associations (MTA) and candidate genes. A total of 147 stable markers associated with the tested 6 agronomic traits were identified. Among them, some MTAs were associated with both heading and anthesis date, and these MTAs were clustered on chromosome 2B. In addition, a total of 21 candidate genes related to the agronomic traits were deduced in introgression lines derived from wild emmer. Among them, the candidate gene associated with 1000-kernel weight on chromosome 7A maybe related to cyclin protein. These MTAs and candidate genes could provide the essential information for cloning genes related to excellentagronomic traits and further provide basis and guidance for comprehensive utilization of wild emmer for genetic improvement in common wheat background.
wild emmer wheat; common wheat; agronomic traits; wide hybridization; GWAS
10.3724/SP.J.1006.2023.21006
本研究由四川省国际科技创新合作项目(2021YFH0110)和四川省生物育种重大科技专项(2022ZDZX0014)资助。
This study was supported by the International Science and Technology Innovation Cooperation Project of Sichuan Province (2021YFH0110) and the Major Science and Technology Project for Biological Breeding of Sichuan Province (2022ZDZX0014).
伍碧华, E-mail: wubihua2017@126.com; 黄林, E-mail: lhuang@sicau.edu.cn
E-mail: jialiu251776931@163.com
2022-01-25;
2022-09-05;
2022-09-28.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20220926.1848.002.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
