基于简化基因组测序技术的喜马拉雅旱獭遗传多样性分析
2023-02-22南新营赵坤钰李耀东
李 优,南新营,赵坤钰,李耀东
(青海大学,西宁,810016)
喜马拉雅旱獭(Marmota himalayana)属啮齿目(Rodentia),松鼠科(Sciuridae),旱獭属(Marmota),是高原环境中的世居动物,也是高原环境中的特有物种,主要分布于我国青海、西藏、云南和青藏高原边缘山地[1]。由于喜马拉雅旱獭长期生活在高原地区,对低氧环境有很好的适应性,在生理生化上获得了适应高原地区低温低氧的稳定遗传学特性,是研究高原低氧适应的天然理想动物模型[2]。目前,对于喜马拉雅旱獭的研究有起源与分化[3]、转录组学[4]、实验动物学[5]和线粒体基因组多态性及群体结构[6]等,对于喜马拉雅旱獭的研究较为成熟。近年来随着分子生物学技术高速发展,基于第二代测序技术的简化基因组测序随之产生并得到广泛应用,其中双酶切RAD(dd-RAD)测序技术是在传统二代测序RAD-Seq 的基础上研发的双酶切RAD-Seq[7],被广泛应用于群体遗传结构和多样性分析[8]以及群体遗传进化[9]等领域,能快速鉴别高密度单核苷酸多态性(SNP)位点[10]。SNP 位点作为基因组中最容易发生变异的类型,可作为群体遗传学和定量遗传学的理想工具[11],但迄今为止,尚未有利用dd-RAD测序技术对喜马拉雅旱獭开展遗传多样性研究的相关报道。本研究基于dd-RAD 测序技术对喜马拉雅旱獭进行群体遗传分析,通过SNP 位点刻画其遗传特性,构建系统进化树,分析种群结构特征,从基因水平分析不同地区个体之间的遗传进化关系,旨在了解青藏高原本地野生动物资源的遗传潜力,为喜马拉雅旱獭比较基因组学和旱獭属动物的资源合理利用提供参考。
1 研究方法
1.1 样本采集
共使用44 例喜马拉雅旱獭样本,按地点分为3个地理种群,其中14例采自青海省玉树州曲麻莱县麻多乡(简称玉树,编号为YS1~YS14;海拔4 452 m;35°2'52″ N,96°37'39″ E),18例采自黄南州同仁县瓜什则乡(简称黄南,编号HN1~HN18;海拔3 273 m;35°49'35″ N,102°28'28″ E),12例采自青海省海东市乐都区引胜乡(简称乐都,编号LD1~LD12;海拔 2 279 m;36°60'19″ N,102°40'37″ E)。取样时喜马拉雅旱獭均已用乙醚深度麻醉处死,符合动物伦理要求,将得到的喜马拉雅旱獭肝脏组织用液氮快速冷冻,放入实验室冰箱-80 ℃贮存备用。
1.2 基因组DNA提取与质量控制
用EasyPure 基因组提取试剂盒(北京全式金生物技术股份有限公司)提取喜马拉雅旱獭基因组DNA,用1.5%琼脂糖凝胶电泳和紫外分光光度计NanoDrop 2000C 仪器检测提取的基因组DNA 质量,基因组DNA 的A260/280值为1.7~2.0符合质量要求,样本基因组DNA于-20 ℃保存备用。
1.3 dd-RAD文库构建
通过参考基因组序列模拟限制性内切酶的酶切位点的数量与分布,选择EcoRⅠ限制性内切酶和NlaⅢ限制性内切酶进行双酶切试验并建库。质检合格的基因组DNA 样本采用dd-RAD 构建文库,具体方法:取喜马拉雅旱獭基因组DNA 1 µg 作为起始量建库,加入10×NE Buffer 5 µL,加入1 µL限制性内切酶EcoRⅠ(NEB)和1 µL 限制性内切酶NlaⅢ,最后加入无酶水使反应体系达到50 µL。反应体系在37 ℃下温育15 min,在65 ℃温育20 min,热失活后进行末端修复,加入接头。使用2%的琼脂糖凝胶电泳对连接产物进行片段选择,选择400~600 bp 的条 带,切胶回收酶切产物。文库质量控制合格后,用 Illumina TruSeq 测序平台对构建的DNA 文库进行简化基因组测序。
1.4 全基因组SNP筛查、分型与质控
用GATK 软件工具包检测SNP[12]。以高质量测序数据的结果为依据,为保证检测得到的SNP 的准确性,用Samtools 去重复,GATK 局部重比对、校正碱基质量值,再使用GATK 进行核苷酸多态性的检测、过滤,得到最终的SNP位点集。质控条件:丢弃低质量(Q<20)和缺失预期限制的位点,同时丢弃具有较小等位基因频率(MAF<0.05)和最大杂合度(He>0.7)的SNP。此外,对每个个体获得的基因型进行支持每个等位基因的reads 数量的询问,丢弃少于20 个reads 支持的基因型和其中一个等位基因的覆盖率比另一个等位基因高3 倍以上的基因型。最后,丢弃所有样本中低于75%个体可以分型的位点。
1.5 数据统计与分析
利用BWA 软件[13]、GATK 软件、ADMIXTURE 软件[14]、GCTA 软件[15]和Fast Tree 软件[16]分别对SNP注释、遗传分化系数统计,并对SNP位点观测杂合度(Ho)、期望杂合度(He)、近交系数(Fis)、多样性指数(Shannon)、多态信息含量(PIC)、有效等位基因数(Ne)和最小等位基因频率(MAF)7 个遗传多样性参数进行分析。基于得到的SNP,使用ADMIXTURE软件计算群体结构,以分群数(K值)为2~10 进行聚类,做出群体结构聚类分析。使用GCTA 软件,通过主成分分析(PCA)获得样本遗传距离远近关系,辅助进化分析。
2 结果
2.1 测序数据统计
基于dd-RAD 测序技术构建44 例喜马拉雅旱獭的测序文库,共获得106 699 757条原始序列,平均每个样本的测序序列为2 424 994 条,获得总碱基数为30 729 530 016 个,平均值为698 398 409 个,喜马拉雅旱獭样本测序获得的GC比例为39.53%~41.09%,平均GC 比例为40.39%,所有样本的Q20是100%,所有的Q30在82.51%以上,表明测序结果较准确,碱基错误率低,数据质量合格,可用于进一步分析(表1)。

表1 喜马拉雅旱獭dd-RAD测序数据Tab.1 dd-RAD sequencing data of Marmota himalayana
2.2 SNP检测与位点开发
44 例喜马拉雅旱獭样本共检测出19 279 845 个SNP 位点。转换SNP 为147 893~332 375,颠换SNP为81 923~186 554,发生转换与颠换的比值为1.759~1.809,转换/颠换均大于1.5,表明喜马拉 雅旱獭与大多数脊椎动物相同,存在转换型颠倒 偏差现象[17]。SNP 遗传多样性分析结果显示,杂合 突变数为21 598~59 268,纯合突变数为208 218~463 280(表2)。
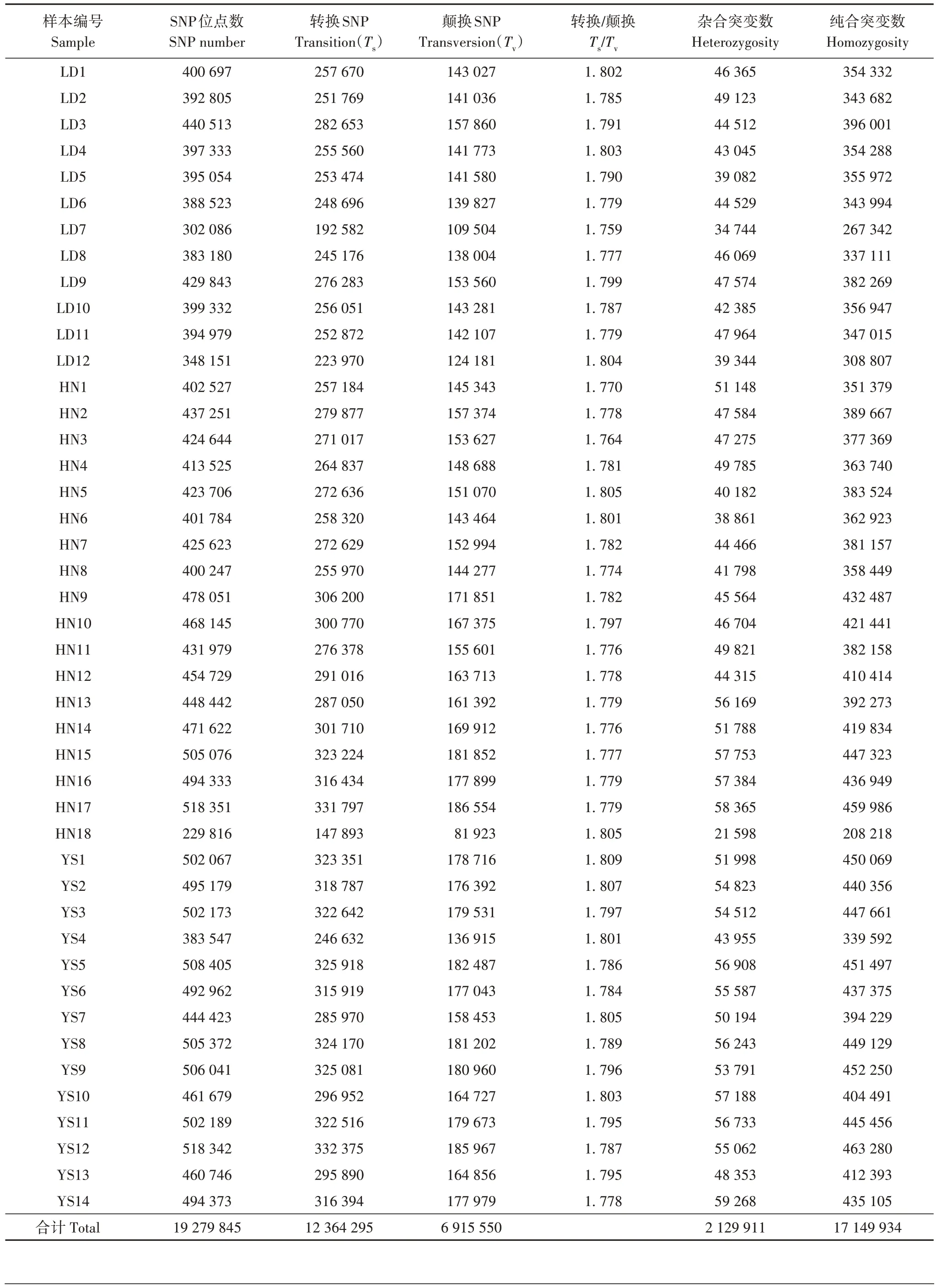
表2 喜马拉雅旱獭SNP统计结果Tab.2 Statistical results of SNP of Marmota himalayana
2.3 遗传进化分析
2.3.1 遗传统计量分析
根据得到的SNP结果,分析遗传统计量,结果显示:乐都(0.236)和玉树(0.269)喜马拉雅旱獭观测杂合度均大于期望杂合度(0.232、0.235),黄南则是期望杂合度(0.254)大于观测杂合度(0.240)。近交系数为0.014~0.145。3 个地区的喜马拉雅旱獭的多态信息含量均小于0.250,为低度多态性。黄南有效等位基因数为1.435,最接近等位基因数2,3 个地区喜马拉雅旱獭有效等位基因数在种群中分布均匀程度从大到小依次为黄南、玉树、乐都。最小等位基因频率为0.183~0.192。3个地区喜马拉雅旱獭Shannon多样性指数为2.855~3.322,从大到小依次为黄南、玉树、乐都(表3)。

表3 遗传统计量信息Tab.3 The results of genetic statistics
2.3.2 群体结构聚类
群体结构分析结果表明,44 例喜马拉雅旱獭遗传背景一致的个体都聚类在一起,说明聚类结果准确。拥有最低交叉验证错误率(CV error)的分群数被认为是最优分群,由图1 可知,交叉验证错误率曲线呈现上升趋势,当分群数(K)为2 时,最低交叉验证错误率值最小,即K=2 为最佳分群数,3 个地区的喜马拉雅旱獭被划分为2 个不同的类群,说明样本之间共享2个基因库。
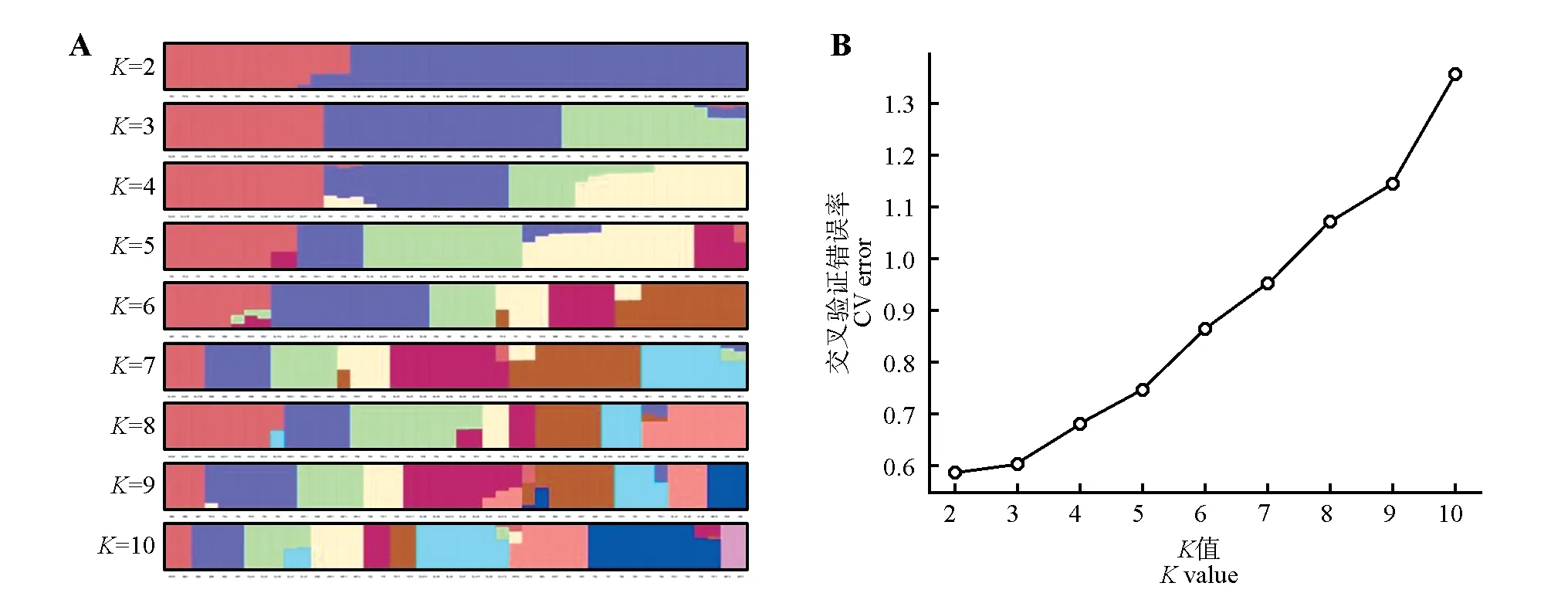
图1 群体结构(A)及不同K值对应的交叉验证错误率(B)Fig.1 Group structure(A)and cross validation error rate corresponding to different K values(B)
2.3.3 PCA分析
主成分分析结果显示,选取的喜马拉雅旱獭样本存在3 个明显的分组,乐都、玉树和黄南的喜马拉雅旱獭各自聚在一起(图2),部分旱獭亲缘关系较为紧密,而部分旱獭亲缘关系较远。
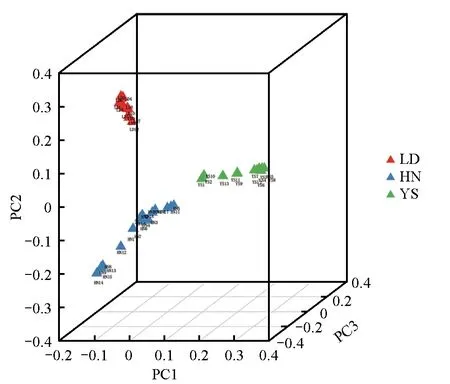
图2 44例喜马拉雅旱獭PCA分析Fig.2 PCA analysis of 44 Marmota himalayana
2.3.4 系统进化分析
系统进化树表明,44 例喜马拉雅旱獭样本汇聚成3 个较大的遗传分支。第1 类、第2 类是黄南的喜马拉雅旱獭,第3类包括乐都和玉树2个地区的喜马拉雅旱獭(图3)。

图3 基于邻接法的44例喜马拉雅旱獭的系统发育树Fig.3 The phylogenetic tree of 44 Marmota himalayana based on adjacency method
3 讨论
青藏高原特有的地理环境使之成为高原自然界的原始“本底”,保存了许多珍稀濒危生物,为开展有关青藏高原生物学、高原医学、动植物遗传多样性及保护等学科的研究,提供了理想的基地和天然实验室[18]。喜马拉雅旱獭作为青藏高原特有物种,成为高原环境相关性研究的理想模型[19]。
本研究利用dd-RAD 测序技术深入了解不同地区喜马拉雅旱獭间的遗传变异,并对其多样性进行分析,青海省3 个地区的喜马拉雅旱獭类群中共鉴定出19 279 845 个SNP 位点,说明喜马拉雅旱獭群体具有丰富的遗传多样性,这些多样性是物种适应环境变化的基础。
从遗传变异指标中可知,乐都和玉树喜马拉雅旱獭的观测杂合度大于期望杂合度,表明这2 个地区的喜马拉雅旱獭杂合过度,黄南喜马拉雅旱獭观测杂合度小于期望杂合度,表明种群内纯合子较多,杂合子缺失[20]。近交系数越大表示基因纯化程度越高,玉树喜马拉雅旱獭近交系数最大。多态信息量可判断等位基因的频率,等位基因越多,多态信息量越大,3个地区中黄南喜马拉雅旱獭的多态信息量最大,相对于其他2 个地区的喜马拉雅旱獭的多态性更高。有效等位基因数可在一定程度上反应种群变异程度,黄南喜马拉雅旱獭有效等位基因数更接近等位基因数,表明群体中的等位基因分布较为均匀。等位基因频率用来表示基因库的丰富程度,黄南的最小等位基因频率最大,表明黄南样本基因库丰富程度高。3 个地区多态信息含量、有效等位基因数、最小等位基因频率和多样性指数(Shannon)大小均呈现相同规律。
从群体结构聚类分析可知,44 例喜马拉雅旱獭分为明显的两群。从PCA 分析可知,44 例喜马拉雅旱獭分成明显的3组,3个地区的喜马拉雅旱獭各自聚类在一起。从系统发育树可见,黄南喜马拉雅旱獭分为2个遗传分支,乐都和玉树样本汇聚成1个大的遗传分支,说明这2 个地区的喜马拉雅旱獭亲缘关系更近,且处在相同地区的喜马拉雅旱獭亲缘关系更近。从地理距离看,黄南和乐都种群的距离较近,但没有完全聚类在一起,没有呈现出明显的系统地理分布格局,同在黄南的喜马拉雅旱獭分为2 个遗传分支的原因可能是青藏高原地势复杂,河流山川众多,以及近年来青海省旅游业发展迅速,人类活动等对喜马拉雅旱獭种群活动范围起到了限制作用。由以上可知,喜马拉雅旱獭的种群进化较复杂,群体结构可能与地理环境有相关性。
综上所述,本研究基于简化基因组测序,构建系统发育树,从基因水平分析不同地区喜马拉雅旱獭个体之间的遗传进化关系,有助于对该物种种群多样性的保护,为高原野生动物疾病的预防提供新的方向和思路。
