玉米雌穗产量相关性状全基因组关联分析与候选基因鉴定
2023-02-10殷芳冰李雅楠鲍建喜马雅杰秦文萱王锐璞李金萍董振营万向元
殷芳冰 李雅楠 鲍建喜 马雅杰 秦文萱 王锐璞 龙 艳, 李金萍 董振营,,* 万向元,,*
玉米雌穗产量相关性状全基因组关联分析与候选基因鉴定
殷芳冰1李雅楠1鲍建喜1马雅杰1秦文萱1王锐璞1龙 艳1,2李金萍2董振营1,2,*万向元1,2,*
1北京科技大学生物与农业研究中心 / 化学与生物工程学院 / 顺德研究生院 / 北京中智生物农业国际研究院, 北京 100083;2北京首佳利华科技有限公司 / 主要作物生物育种北京市工程实验室 / 生物育种北京市国际科技合作基地, 北京 100192
玉米雌穗产量相关性状直接影响玉米最终产量, 解析其遗传机制可为玉米高产提供有益指导。本研究以733份玉米自交系作为关联群体, 在2个环境下随机区组种植, 调查穗行数(KRN)、穗长(EL)和穗粗(ED) 3个产量相关性状, 利用MaizeSNP3072芯片对其进行基因分型, 采用FarmCPU模型进行全基因组关联分析, 分别鉴定出16、13和24个与3个性状显著关联的单核苷酸多态性位点(SNP), 对表型变异的解释率分别为0.01%~7.08%、0.01%~5.34%和0.07%~4.34%。其中, 分别有6、2和5个与3个性状存在显著关联的高可信度(high confidence, HC) SNP, 而且有2个HC-SNP同时与KRN和ED显著相关, 1个KRN HC-SNP和3个ED HC-SNP为本研究首次报道。在所鉴定HC-SNP上下游200 kb范围内筛选出33个重要候选基因, 其中9号染色体SNP标记PZE-109003046所在基因为控制生长素极性运输从而调控雌穗性状的已知基因。另一些候选基因编码不同转录因子, 以及参与生长素、赤霉素和乙烯等激素介导的信号转导、DNA甲基化和蛋白磷酸化等翻译后修饰过程的蛋白, 可能从不同方面调控雌穗相关性状。本研究所挖掘的11个HC-SNP与33个候选基因可以为进一步克隆雌穗性状功能基因、揭示相关分子调控机制以及利用分子标记辅助选择育种提供有益指导。
玉米; 雌穗产量相关性状; 全基因组关联分析; 候选基因
玉米雌穗是重要的生殖器官, 也是产量形成要素的决定因子之一。玉米雌穗性状属于典型多基因控制的数量性状, 其遗传力较高, 穗行数(kernel row number, KRN)、穗长(ear length, EL)、穗粗(ear diameter, ED)等各子性状之间存在相关性, 共同影响玉米产量[1]。因此, 解析玉米雌穗性状遗传结构和挖掘雌穗性状重要调控位点和基因对提高玉米产量具有重要意义。
随着分子标记与高通量测序技术的快速发展, QTL (quantitative trait locus)定位和全基因组关联分析(genome-wide association study, GWAS)已成为解析玉米雌穗性状遗传机制的重要方法, 目前利用2种方法已经定位出多个调控穗行数、穗长、穗粗等性状的遗传位点[2-4]。如Li等[5]利用农系531和H21为亲本构建的染色体片段代换系(chromosome segment substitution lines, CSSL)和691个SSR分子标记,共检测到11个调控穗行数的QTL位点, 其中单个QTL可解释9.87%~19.44%的表型变异。Choi等[6]使用HF1和11S6169构建的双单倍体(doubled haploid, DH)群体进行QTL定位, 共鉴定出2个调控穗长和1个调控穗粗的QTL位点, 解释表型变异的12.48%~38.08%。Zhou等[7]利用F2:3群体、四交群体和关联群体对穗长性状进行QTL定位和GWAS分析,共鉴定出14个QTL位点和4个SNP位点。Zhang等[8]以B73×Mo17 (IBM) Syn10 DH群体和关联群体为材料, 利用QTL定位和GWAS方法, 分别鉴定出11、13和21个控制穗粗、穗长和穗行数的QTL位点以及16、13和14个与穗粗、穗长和穗行数显著关联的SNP位点。Xu等[9]以339个玉米自交系作为关联群体, 利用GEMMA (genome-wide efficient mixed model association)和LASSO (least absolute shrinkage and selection operator)算法进行GWAS分析, 共鉴定出3个与穗行数显著关联的SNP位点, 单个SNP位点可解释0.40%~9.69%的表型变异。Li等[10]利用121份玉米自交系组成的关联群体进行GWAS分析, 在2个环境共鉴定出84个与穗长显著关联的SNP位点, 其中5个SNP位点在2个环境中均能检测到。这些研究为解析玉米雌穗性状的遗传结构奠定了重要基础。
虽然目前已有雌穗性状遗传位点挖掘相关研究开展, 但是各研究所选用的群体、标记及分析方法不同, 所鉴定出的位点也存在显著差异, 因此仍需进一步深入开展玉米雌穗性状遗传结构研究。本研究选取具有广泛表型多样性的733份玉米自交系作为关联群体, 利用MaizeSNP3072芯片[11]基因型数据对玉米穗行数、穗长和穗粗性状展开全基因组关联分析, 深入挖掘调控上述雌穗性状的重要遗传位点并鉴定其优异等位变异, 为利用分子标记辅助选择进行玉米雌穗性状遗传改良及精细定位与克隆雌穗性状关键基因提供理论支持。
1 材料与方法
1.1 试验材料与田间设计
试验材料为由北京科技大学生物与农业研究中心所保存的包含733份表型变异较为丰富的玉米自交系。分别在2019年夏和2020年夏种植于北京平谷, 采用随机区组设计, 每个自交系种植2行, 行长6 m, 行距0.6 m, 株距0.2 m, 每行30株, 种植期间按照正常的农业生产进行田间管理。
1.2 雌穗性状表型测定与数据统计分析
植株生理成熟后, 在每份玉米自交系穗行中间随机收取5个果穗[9,12-14], 利用自动化果穗考种仪(YTS-MET, 谷丰光电)进行穗行数、穗长和穗粗等雌穗性状测量。将采集到的表型数据利用SPSS软件进行描述性统计分析和联合方差分析, 利用R语言“PerformanceAnalytics”软件包进行频率直方图分析和相关性分析, 利用R语言“lme4”软件包计算广义遗传力和最佳线性无偏预测值(best linear unbiased prediction, BLUP)。
1.3 基因型鉴定和分析
利用MaizeSNP3072芯片[11]对733份玉米自交系进行基因型鉴定, 利用PLINK软件去除最小等位基因频率(minor allele frequency, MAF) < 0.05, 缺失率(missing rate) > 20%的位点, 最终筛选出2799个高质量SNP用于后续全基因组关联分析。
1.4 亲缘关系与主成分分析
利用Tassel 5.0软件Centered_IBS方法进行亲缘关系计算, 使用R语言“heatmap”软件包进行亲缘关系图绘制。利用PLINK软件进行主成分分析(principal components analysis, PCA), 并使用R语言“ggplot2”软件包进行PCA图绘制。
1.5 全基因组关联分析
使用R语言“GAPIT”软件包的FarmCPU模型, 在考虑研究材料群体结构与亲缘关系因素下进行SNP标记与穗行数、穗长和穗粗的关联分析。在< 0.0001的水平上, 判定SNP标记与穗行数、穗长和穗粗关联的显著性, 显著标记的表型变异解释率(phenotypic variance explained, PVE)利用线性回归的方法进行计算[15]。
1.6 候选基因预测
根据MaizeGDB数据库(http://www.maizegdb. org/gbrowse) B73参考基因组(B73 RefGenv4)序列信息, 获取显著关联SNP上下游200 kb范围内基因作为玉米雌穗性状候选基因。在Uniprot蛋白质数据库(https://www.uniprot.org/uniprot)对候选基因进行注释及功能预测。
2 结果与分析
2.1 玉米雌穗性状表型统计分析
通过调查2个环境表型数据, 发现穗行数在2019年和2020年变异范围分别为8.00~21.33和8.40~24.40, 穗长变异范围分别为6.78~21.83 cm和8.34~21.03 cm, 穗粗变异范围分别为3.11~5.67 cm和3.09~5.83 cm (表1)。表明本关联群体穗行数、穗长和穗粗表型变异范围较大, 表型多样性较为丰富。各性状偏度和峰度绝对值均小于1 (表1), 结合频率直方图(图1), 显示3个性状均呈现正态分布,符合典型的数量性状特征。各性状表型数据在不同年份间呈现显著相关(= 0.48~0.67,< 0.001), 同时发现穗行数和穗粗之间呈现正向显著相关(= 0.39~0.63,< 0.001) (图1)。穗行数、穗长和穗粗遗传力分别为0.80、0.65和0.79 (表1), 联合方差分析显示关联群体基因型间存在极显著差异, 不同环境间和基因型与环境互作也存在极显著差异(表2)。表明尽管雌穗性状遗传力较高, 但也受部分环境因素的影响, 其中环境因素对穗长表型影响最大。
2.2 亲缘关系与主成分分析
利用PLINK进行主成分分析, 发现733份玉米自交系主要分为3个组分, 基本上对应了玉米自交系常见的3个亚群, 即分别为以自交系B73为代表的坚秆群体(stiff stick, SS), 自交系郑58 (Zheng 58)为代表的非坚秆群体(non-stiff stick, NSS), 自交系昌7-2 (Chang 7-2)为代表的黄早四群体(HZS) (图2-a)。前2个主成分能够清晰的区分3个亚群, 第1个主成分反映了SS和HZS的分化, 第2个主成分反映了NSS和SS或HZS的分化。因此后续利用主成分分析产生的Q矩阵作为协变量进行关联分析。利用Tassel进行kinship计算发现大部分材料亲缘关系系数为–0.5~0.5, 表明无明显亲缘关系或存在较弱的亲缘关系; 仅有极少部分材料亲缘关系系数为0.5~1.0 (图2-b), 后续利用亲缘关系分析计算出的K矩阵作为协变量进行关联分析。
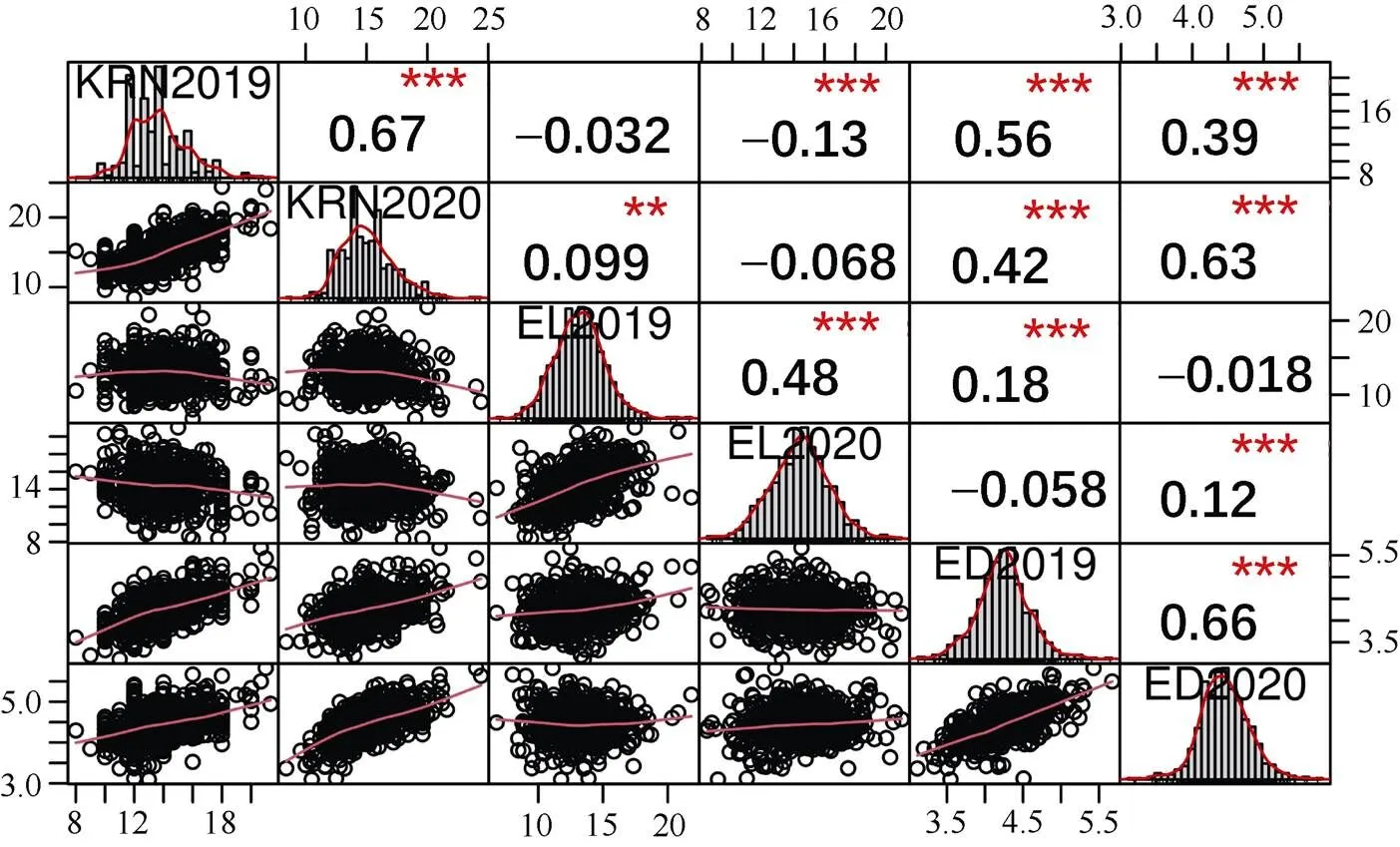
图1 不同环境间穗行数、穗长和穗粗相关性分析和频率直方图
图中对角线表示穗行数、穗长和穗粗在2019年北京和2020年北京环境的频率直方图, 左下表示穗行数、穗长和穗粗在不同环境下的散点图, 右上表示相关性系数。**、***分别表示在< 0.01、0.001水平上差异显著。KRN、EL和ED分别表示穗行数、穗长和穗粗。
The diagonal line and lower left show the frequency histogram and scatter diagram of KRN, EL, and ED traits in different environments, respectively. The upper right shows the correlation coefficients between different traits.**and***indicate significance at< 0.01 and< 0.001, respectively.KRN, EL, and ED represent kernel row number, ear length, and ear diameter, respectively.
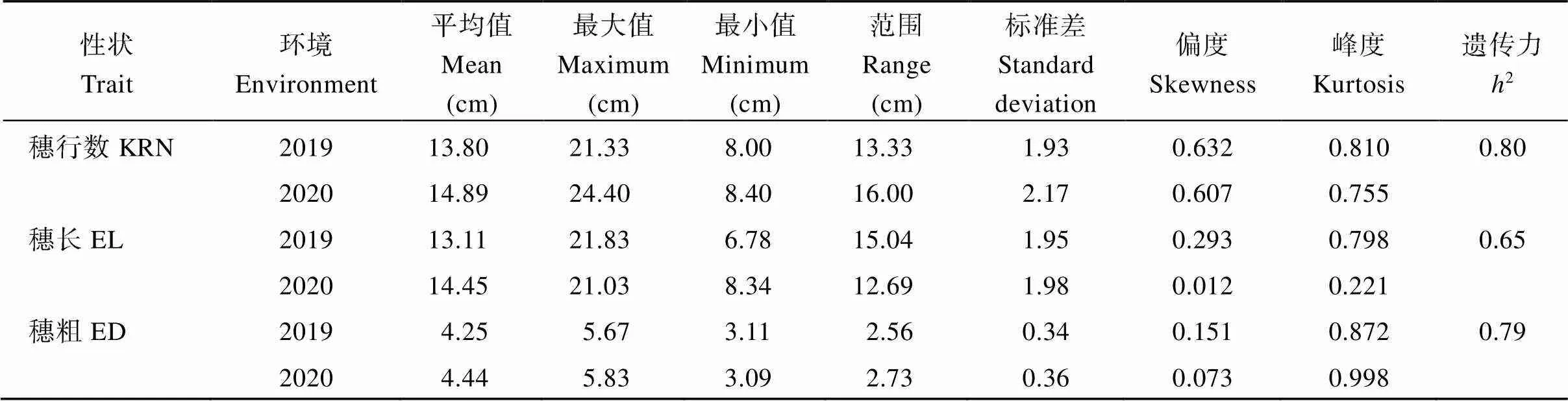
表1 穗行数、穗长和穗粗表型数据统计分析
KRN、EL和ED处理缩写同图1。
Abbreviations of KRN, EL, and ED are the same as those given in Fig. 1.
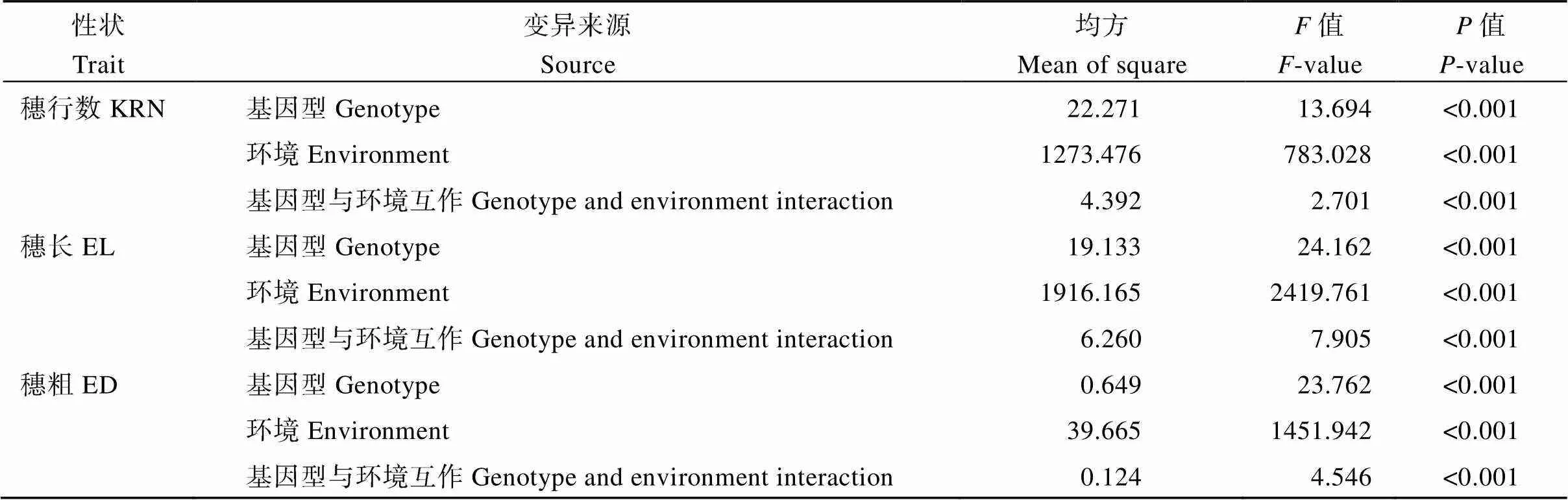
表2 穗行数、穗长和穗粗多环境联合方差分析
KRN、EL和ED处理缩写同图1。
Abbreviations of KRN, EL, and ED are the same as those given in Fig. 1.
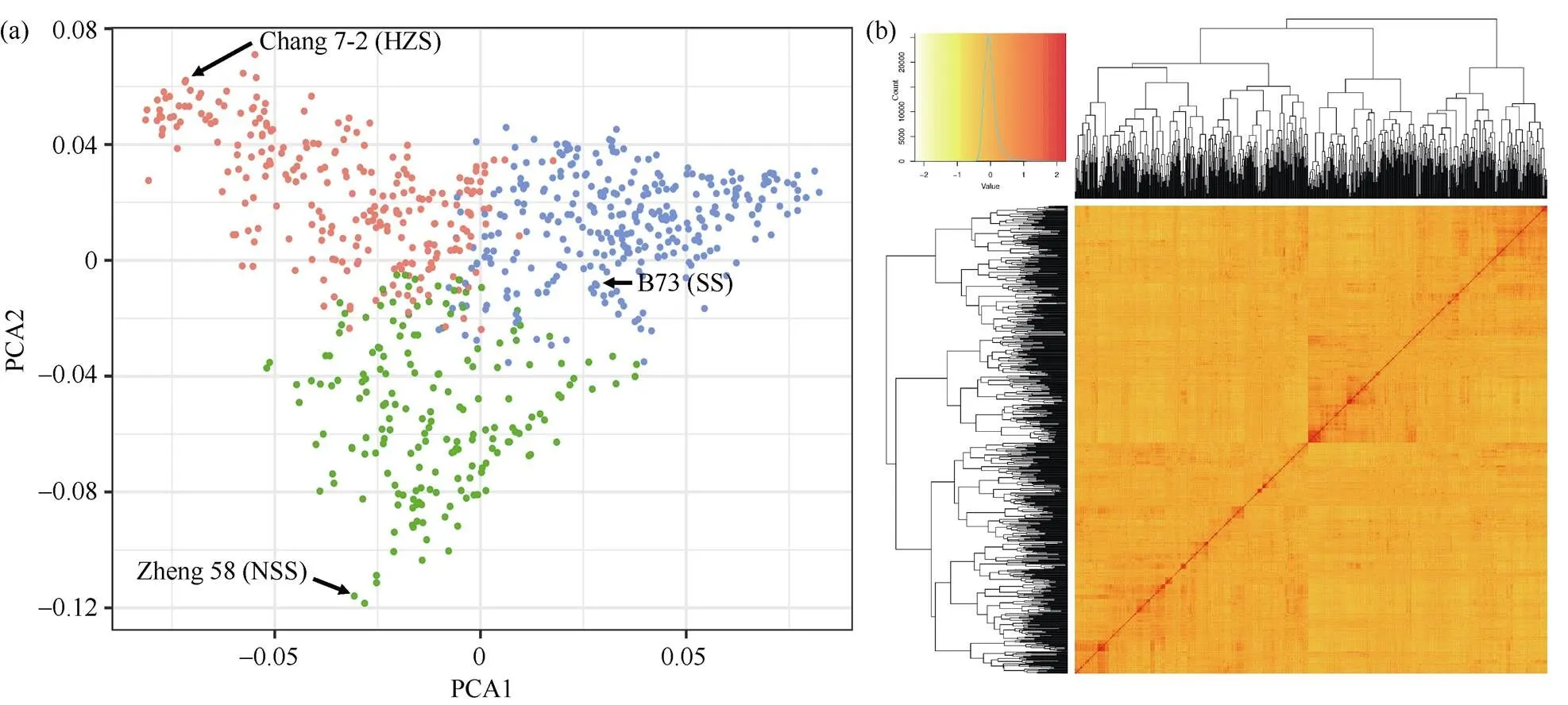
图2 733份玉米自交系主成分分析(a)与亲缘关系分析(b)
HZS、NSS和SS分别表示黄早四群体、非坚秆群体和坚秆群体。
HZS, NSS, and SS represent the heterotic groups of Huangzaosi, non-stiff stick, and stiff stick, respectively.
2.3 玉米雌穗性状全基因组关联分析
利用2799个SNP标记分别对2019年和2020年穗行数、穗长、穗粗表型数据及其BLUP值进行全基因组关联分析, QQ-plot显示关联分析所选取的统计模型较为合理, 分别检测到16、13和24个与穗行数、穗长、穗粗性状显著关联的SNP位点(图3和附表1)。穗行数表型显著关联SNP位点分布在玉米8条染色体, 在4号、6号和8号染色体各检测到3个, 在7号和9号染色体各检测到2个, 在3号、5号和10号染色体各检测到1个; 单个位点表型变异解释率为0.01%~7.08%, 其中表型解释率最高的位点为4号染色体的PZE-104126211 (附表1)。穗长表型显著关联SNP位点分布在玉米6条染色体,在8号染色体检测到最多, 为4个; 其次为4号染色体, 为3个; 在2号和5号染色体各检测到2个, 在1号和6号染色体各检测到1个; 单个位点表型变异解释率为0.01%~5.34%, 其中表型解释率最高的位点为8号染色体的PZE-108133100 (附表1)。穗粗表型显著关联SNP位点在玉米10条染色体均有分布, 其中4号染色体最多, 为6个, 在1号、3号、6号、8号和10号染色体分布最少, 均为1个; 单个位点表型变异解释率为0.07%~4.34%, 表型解释率最高的位点为3号染色体的PZE-103087199 (附表1)。

图3 玉米穗行数、穗长、穗粗GWAS曼哈顿图(a, c, e)与QQ-plot图(b, d, f)
BLUP表示最佳线性无偏预测值。KRN、EL和ED处理缩写同图1。
BLUP represents best linear unbiased prediction. Abbreviations of KRN, EL, and ED are the same as those given in Fig. 1.
进一步分析发现可被2次及以上独立关联分析检测到的穗行数、穗长和穗粗显著关联SNP分别为6、2和5个, 这些SNP针对相应性状可被多次显著关联, 属于高可信度(high confidence, HC) SNP, 其中HC-SNP标记PZE-107116723在所有3次独立穗行数关联分析中均显著关联, 且该位点为本研究首次报道(表3)。3个HC-SNP位点, 即4号染色体PZE-104093153、PZE-104126211和5号染色体SYN32729同样为本研究首次报道与穗粗显著相关。4号染色体HC-SNP位点PZE-104126211和6号染色体HC-SNP位点SYN4194与穗行数和穗粗性状同时显著关联, 属于一因多效位点。PZE-104126211是本研究所检测对穗行数表型贡献率最高的位点, 为7.08%, 该位点对穗粗的表型贡献率为0.66%; SYN4194位点在穗粗中的表型贡献率为2.66%, 在穗行数中的表型贡献率为0.72% (表3)。
综上, 本研究关联到11个与雌穗性状显著关联的HC-SNP, 其中有2个HC-SNP与穗行数和穗粗同时显著关联, 并鉴定出1个穗行数和3个穗粗HC-SNP新位点(表3)。
2.4 候选基因分析
从以上11个与穗行数、穗长和穗粗显著关联的HC-SNP位点上下游200 kb范围内进行候选基因筛选, 结合基因功能注释和前人文献报道, 共鉴定出33个重要候选基因(表3)。经统计, 33个候选基因中8个为转录因子编码基因, 3个为生长素相关基因(表3)。其中穗粗性状显著关联SNP位点PZE-109003046候选基因()为编码PIN蛋白的调控雌穗性状的已知基因, 在生长素极性运输中发挥重要作用[16-17]。与穗行数和穗粗性状均显著关联的SYN4194位点携带编码Auxin response factor (ARF)的候选基因, 穗行数和穗粗均显著关联位点PZE-104126211候选基因编码C2H2锌指蛋白(C2H2-like zinc finger protein), 穗粗显著关联SNP位点SYN32729候选基因编码bHLH转录因子(Transcription factor bHLH157), 暗示这些已被大量研究证明在植物生长发育中发挥重要作用的转录因子可能对雌穗发育同样具有重要调控作用。
同时发现一些候选基因编码表观遗传修饰、蛋白翻译后修饰等相关蛋白。如穗行数显著关联SNP位点PZA03608.2候选基因编码DNA甲基转移酶(DNA methyl transferase)。调控穗行数和穗粗的SYN4194位点候选基因编码非特异性丝氨酸/苏氨酸蛋白激酶。
2.5 重要位点优异等位变异鉴定和表型效应分析
PZE-107116723为本研究所鉴定穗行数相关HC-SNP新位点, 在穗行数性状所有独立分析中均被显著关联, 推测该位点携带可在不同环境下稳定调控穗行数的重要基因。在本研究自交系群体中, 151份材料在该位点携带A/A基因型, 545份材料在该位点携带G/G基因型, 其余37份材料在该位点基因型缺失。发现该位点携带A/A基因型材料比携带G/G基因型材料2个年份穗行数分别平均增加0.86行和1.04行, 差异达均到极显著水平(<0.001), 因此鉴定A/A基因型是该位点的优异等位变异, 该等位变异对穗长和穗粗性状没有显著影响(图4-a)。PZE-104093153、SYN32729和PZE-104126211为本研究首次鉴定与穗粗显著关联的HC-SNP位点, 发现PZE-104093153位点A/A优异等位变异仅显著增加穗粗(图4-b), SYN32729位点G/G优异等位变异则同时显著提高穗行数(图4-c), 表明SYN32729位点可能是潜在的一因多效位点。HC-SNP标记PZE-104126211和SYN4194与穗行数和穗粗性状均显著关联(图3和表3), PZE-104126211位点携带A/A优异等位变异材料比携带G/G等位变异材料的穗行数性状在2个年份分别增加0.86行和1.14行, 差异均达到极显著水平; 穗长分别增加0.11 cm和0.54 cm, 在2020年差异达到极显著水平; 穗粗则在2个环境中分别增加0.01 cm和0.06 cm, 在2020年差异达到显著水平(图4-d)。SYN4194位点G/G优异等位变异分别比A/A等位变异的穗粗在2个环境中分别增加0.12 cm和0.10 cm, 差异均达到极显著水平; 穗长在2个环境中分别增加0.45 cm和0.39 cm, 差异均达到显著水平; 穗行数分别增加0.35行和0.34行, 在2019年环境差异达到显著水平(图4-e)。表明PZE-104126211和SYN4194位点可能携带同时对穗行数、穗长、穗粗均具有调控作用的重要基因。
3 讨论
3.1 雌穗性状GWAS比较分析与重要位点优异等位变异鉴定
本研究利用2799个SNP标记和733份玉米自交系雌穗性状进行全基因组关联分析, 共鉴定出16 个与穗行数、13个与穗长和24个与穗粗性状显著关联的SNP位点, 表型变异解释率分别为0.01%~7.08%、0.01%~5.34%和0.07%~4.34%, 与马娟等[18]结果较为相似, 表明微效多基因调控可能是雌穗性状遗传机制之一。其中11个位点在多次独立关联分析分别与相应雌穗性状显著关联, 属于HC-SNP; HC-SNP位点PZE-104126211和SYN4194则同时与穗行数和穗粗性状显著关联, 属于一因多效位点(表3)。
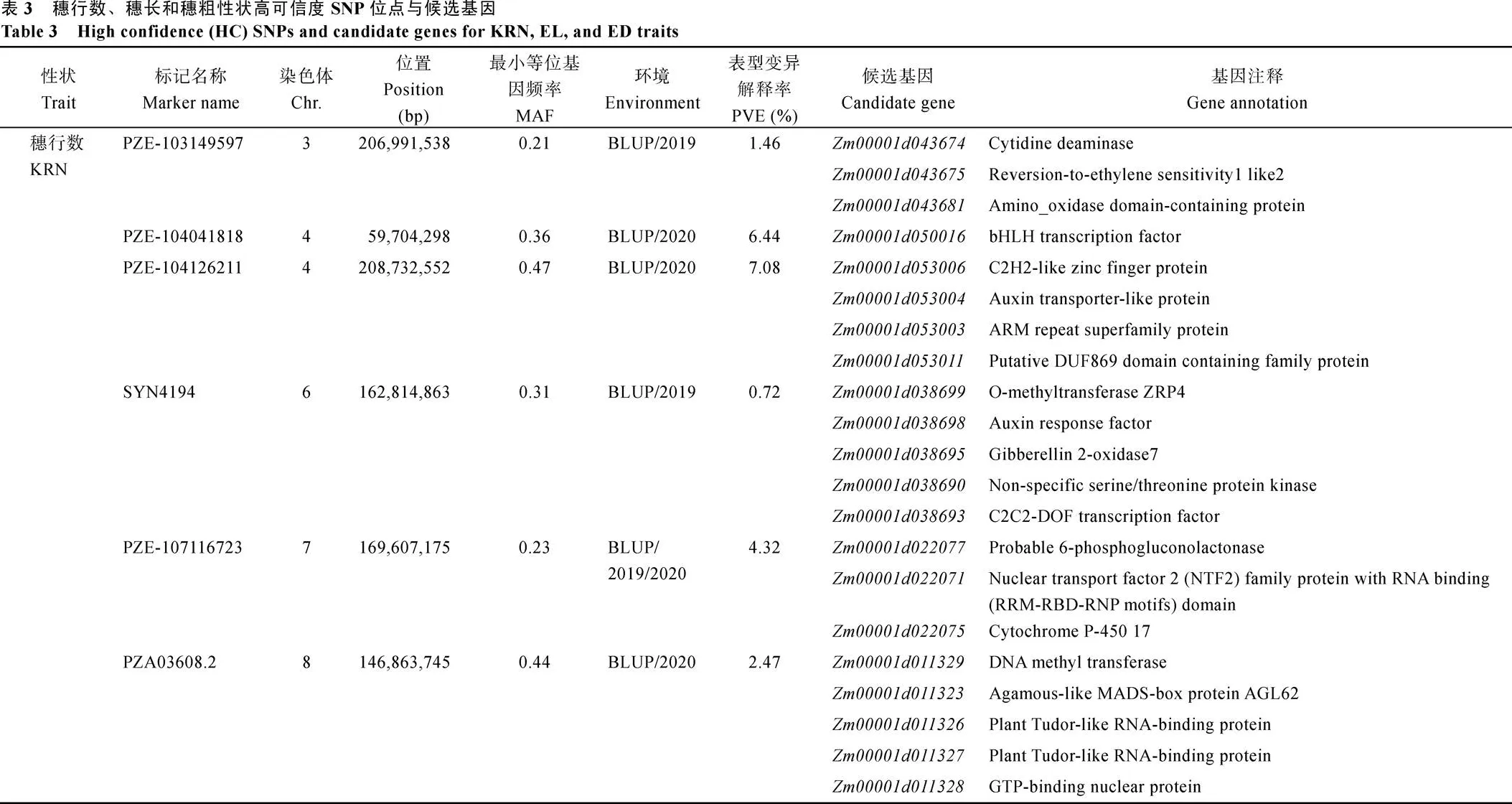
图4 显著关联SNP位点等位变异表型效应分析
Fig. 4 Phenotypic effect analysis of the allelic variation in associated loci
NS表示> 0.05,*、**和***分别表示0.05、0.01和 0.001水平差异显著。KRN、EL和ED处理缩写同图1。
NS:> 0.05.*,**, and***indicate significance at the 0.05, 0.01, and 0.001 probability levels, respectively. Abbreviations of KRN, EL, and ED are the same as those given in Fig. 1.
在前期研究中, 我们通过系统总结国内外雌穗性状遗传定位研究进展, 鉴定出了雌穗性状QTL和SNP热点区间[19]。在6个与穗行数显著关联的HC-SNP位点中, 7号染色体PZE-107116723位点为本研究首次鉴定, 其余5个位点位于已知穗行数QTL区间, 其中4个位点位于我们所鉴定穗行数QTL定位热点区间[19]。如3号染色体206.99 Mb的PZE-103149597同时分别位于Lu等[20]、Karen等[21]和Tian等[22]所定位的穗行数QTL bnlg197– umc1844、umc1659–umc1320及umc1135–umc1140区间, 同时也位于我们所鉴定穗行数QTL热点区间3号染色体203.609~210.466 Mb[19]。4号染色体59.70 Mb的PZE-104041818与Liu等[23]定位的穗行数QTL umc1117–umc1791区间、Ma等[24]定位的穗行数QTL phi079–umc1896区间相吻合, 同时也位于穗行数QTL热点区间4号染色体46.784~64.228 Mb[19]。4号染色体208.73 Mb的PZE-104126211位点位于已知穗行数QTL umc2188–umc1101[20]、bnlg2162–umc1284[23]、umc1371–bnlg1023[25]、umc2286–umc1503[25]、umc1173–umc2286[26]区间内, 并且位于穗行数QTL热点区间4号染色体200.053~208.887 Mb内[19]。SYN4194位于6号染色体162.81 Mb, 与Brown等[27]通过GWAS关联到的穗行数相关SNP位点仅相差约15 kb; 位于Liu等[23]定位的穗行数QTL umc1020– umc1296和umc1859–umc1248区间、Huo等[28]定位的穗行数QTL umc1805–umc1296区间内, 也位于我们所鉴定穗行数QTL热点区间6号染色体158.522~ 166.027 Mb内[19]; PZA03608.2位于8号染色体146.86 Mb, 与Liu等[23]定位的穗行数QTL bnlg2181– umc2199相吻合。这些位点可被不同研究反复验证, 暗示所关联区间携带可在不同环境稳定调控雌穗性状的重要基因。
2个与穗长显著关联的HC-SNP位点均位于已定位穗长QTL区间, 如1号染色体208.33 Mb的SNP位点PZE-101162306位于Zhou等[7]定位的穗长QTL umc1590–bnlg1556区间。4号染色体193.37 Mb的SNP位点PZE-104113905则位于Karen等[21]定位的穗长QTL bnlg2291–umc1989区间内。5个与穗粗显著关联的SNP位点中, 6号染色体162.81 Mb的SNP位点SYN4194与Zhang等[8]和Yang等[29]通过GWAS鉴定的穗粗显著关联SNP位点物理距离分别为0.45 Mb和 0.47 Mb。9号染色体3.29 Mb的SNP位点PZE-109003046存在已知基因。虽然PZE-104126211位于穗行数QTL热点区间[19], 但尚无与穗粗性状共定位的报道, 因此与PZE- 104093153和SYN32729同属于本研究所首次鉴定穗粗性状显著关联位点。
挖掘重要遗传位点优异等位变异是利用分子标记辅助选择育种的首要条件之一。本研究对所关联新位点及一因多效位点进行表型效应分析, 发现携带不同等位变异材料间表型差异显著, 进而鉴定出了相应位点优异等位变异(图4)。我们同时发现一些位点可能携带可以协同改良玉米雌穗性状的关键基因, 如HC-SNP标记PZE-104126211和SYN4194与穗行数和穗粗性状均显著关联, 表型效应分析发现2个位点均同时显著影响穗长性状(图4-d, e), 选择其A/A和G/G优异等位变异有望同时对穗行数、穗长和穗粗性状进行协同改良。GWAS显示SYN32729与穗粗显著关联(图3), 其优异等位变异G/G可同时显著提高穗行数(图4-c), 表明利用该位点可对穗粗和穗行数性状进行协同改良。
3.2 候选基因鉴定与分析
在前期研究中, 我们系统总结了调控玉米雌穗性状的重要已知基因和分子作用机制, 发现CLAVATA-WUSCHEL负反馈循环信号途径、RAMOSA途径、激素合成与代谢及一些重要转录因子和蛋白激酶对雌穗发育发挥重要作用[19]。我们结合数据库基因功能注释、文献分析和雌穗基因表达分析, 在本研究所挖掘11个与穗行数、穗长和穗粗显著关联的HC-SNP位点上下游200 kb区间鉴定出33个重要候选基因(表3)。与穗粗显著关联的9号染色体PZE-109003046位点候选基因为调控雌穗性状的已知基因, 该基因编码的PIN蛋白为生长素运输的载体组分, 在生长素极性运输中发挥重要作用,突变导致雌穗小花减少、畸形甚至秃秆[16-17]。本研究关联到携带雌穗性状重要已知调控基因的SNP位点, 表明关联分析结果较为可靠。
目前所鉴定与雌穗发育相关激素调控基因主要与生长素相关, 如生长素合成相关基因()[30]和()[31]、生长素运输和信号转导相关基因()[32]和()[33-34]等。本研究亦同时在多个位点鉴定到生长素相关基因。如与穗行数和穗粗性状均显著关联的4号染色体PZE-104126211位点候选基因编码Auxin transporter-like蛋白, 同时该位点对穗行数表型解释率最高; 与穗行数和穗粗性状均显著关联的6号染色体SYN4194位点候选基因编码Auxin response factor (ARF), 该类蛋白可与生长素反应启动子元件(AuxRE)结合进而调控下游基因表达[35]。此外, SYN4194位点同时携带编码赤霉素合成相关基因, 3号染色体PZE-103149597位点携带编码乙烯信号响应相关基因。已有报道显示赤霉素和乙烯均可调控玉米雌穗性状[19], 推测上述2个基因可能在玉米雌穗发育中发挥重要作用。
转录因子是一类能与基因5'端上游特定序列专一性结合, 可调控下游基因在特定时空以特定强度表达的蛋白分子。已报道有多个转录因子编码基因与雌穗性状相关, 如编码bHLH转录因子的()基因可以协同调节生长素合成和运输相关基因, 进而调控玉米腋生分生组织和侧生器官的形成和玉米雌穗的发育[36]。C2H2锌指蛋白编码基因()则为调控玉米花序分枝的RAMOSA途径中关键成员,在初生分生组织基部附近的边界区表达, 影响SPM的确定性,突变后雌穗产生额外的长分枝[37]。在本研究33个候选基因中8个为转录因子编码基因, 其中4号染色体穗行数候选基因与5号染色体穗粗候选基因均编码bHLH转录因子, 穗行数和穗粗候选基因编码C2H2锌指蛋白。穗粗候选基因编码AT-hook基序核定位蛋白, 此类蛋白可以与富含AT碱基的DNA序列的结合来行使功能, 对植物生长发育具有重要的调控作用[38]。在雌穗形成过程中发挥重要作用的已知基因()即编码AT-hook蛋白, 该基因突变导致雌穗停止发育或果穗较短, 穗行排列不规则, 因而对玉米雌穗性状产生显著影响[39]。
一些具有蛋白翻译后修饰功能的蛋白编码基因与雌穗发育紧密相关, 如调控花序分生组织发育的[33-34]和调控行粒数的()[40]均编码丝氨酸/苏氨酸蛋白激酶。本研究调控穗行数和穗粗位点SYN4194候选基因编码丝氨酸/苏氨酸蛋白激酶, 穗长调控位点PZE-104113905候选基因编码注释为含蛋白质激酶结构域的蛋白, 推测这些基因可能对雌穗性状同样具有重要调控作用。
4 结论
通过对玉米雌穗产量相关穗行数、穗长和穗粗性状全基因组关联分析, 共鉴定出48个显著关联SNP位点, 其中11个为多次独立关联分析共定位的HC-SNP位点。其中穗行数显著关联HC-SNP位点PZE-107116723、穗粗显著关联HC-SNP位点PZE- 104093153、PZE-104126211和SYN32729为本研究所首次发现, 这些新位点的挖掘有助于进一步揭示玉米雌穗性状的遗传结构。HC-SNP位点PZE- 104126211和SYN4194同时与穗行数和穗粗表型显著关联, 属于一因多效位点, 这些位点可能是雌穗产量相关性状协同改良的重要位点。通过评估所鉴定新位点和一因多效位点的遗传效应, 鉴定出的相应优异等位变异可为玉米雌穗性状遗传改良及高产分子育种提供有益指导。我们同时发现10个HC-SNP位点尚无雌穗性状相关基因克隆报道, 且部分位点位于我们前期研究所鉴定雌穗性状遗传定位热点区间, 这些位点可被不同研究所同时定位, 暗示相应定位区间携带可在不同环境稳定调控雌穗性状的重要基因, 是今后基因克隆和开发分子标记进行辅助育种的重点区间。本研究进一步鉴定出了33个重要候选基因, 其中1个为调控雌穗性状的已知基因, 其他候选基因编码不同转录因子, 以及参与生长素、赤霉素和乙烯等激素介导的信号转导、DNA甲基化和蛋白磷酸化等翻译后修饰过程的蛋白, 暗示这些基因可能从不同方面调控雌穗相关性状。综上, 本研究所鉴定重要遗传位点和候选基因为进一步克隆雌穗产量相关性状功能基因、揭示相关分子调控机制以及分子育种均具有重要指导意义。
[1] 宁慧云, 连晋, 赵玉坤, 连吉明, 高根来. 不同玉米品种雌穗性状及产量的灰关联评价研究. 农学学报, 2013, 3(6): 13–16.
Ning H Y, Lian J, Zhao Y K, Lian J M, Gao G L. Grey relational evaluation study in ear traits and yield of different maize varieties., 2013, 3(6): 13–16 (in Chinese with English abstract).
[2] 王帮太, 吴建宇, 丁俊强, 席章营. 玉米产量及产量相关性状QTL的图谱整合. 作物学报, 2009, 35: 1836–1843.
Wang B T, Wu J Y, Ding J Q, Xi Z Y. Map integration of QTLs for grain yield and its related traits in maize., 2009, 35: 1836–1843 (in Chinese with English abstract).
[3] 吴律, 代力强, 董青松, 施婷婷, 王丕武. 玉米行粒数的全基因组关联分析. 作物学报, 2017, 43: 1559–1564.
Wu L, Dai L Q, Dong Q S, Shi T T, Wang P W. Genome-wide association analysis of kernel number per row in maize., 2017, 43: 1559–1564 (in Chinese with English abstract).
[4] 张焕欣, 翁建峰, 张晓聪, 刘昌林, 雍洪军, 郝转芳, 李新海. 玉米穗行数全基因组关联分析. 作物学报, 2014, 40: 1–6.
Zhang H X, Weng J F, Zhang X C, Liu C L, Yong H J, Hao Z F, Li X H. Genome-wide association analysis of kernel row number in maize., 2014, 40: 1–6 (in Chinese with English abstract).
[5] Li F, Jia H T, Liu L, Zhang C X, Liu Z J, Zhang Z X. Quantitative trait loci mapping for kernel row number using chromosome segment substitution lines in maize., 2014, 13: 1707–1716.
[6] Choi J K, Sa K J, Park D H, Lim S E, Ryu S H, Park J Y, Park K J, Rhee H I, Lee M, Lee J K. Construction of genetic linkage map and identification of QTLs related to agronomic traits in DH population of maize (L.) using SSR markers., 2019, 41: 667–678.
[7] Zhou B, Zhou Z J, Ding J Q, Zhang X C, Mu C, Wu Y B, Gao J Y, Song Y X, Wang S W, Ma J L, Li X T, Wang R X, Xia Z L, Chen J F, Wu J Y. Combining three mapping strategies to reveal quantitative trait loci and candidate genes for maize ear length., 2018, 11: 170107.
[8] Zhang X X, Guan Z R, Li Z L, Liu P, Ma L L, Zhang Y C, Pan L, He S J, Zhang Y L, Li P, Ge F, Zou C Y, He Y C, Gao S B, Pan G T, Shen Y O. A combination of linkage mapping and GWAS brings new elements on the genetic basis of yield-related traits in maize across multiple environments., 2020, 133: 2881–2895.
[9] Xu Y, Xu C, Xu S. Prediction and association mapping of agronomic traits in maize using multiple omic data., 2017, 119: 174–184.
[10] Li T, Qu J Z, Tian X K, Lao Y H, Wei N N, Wang Y H, Hao Y C, Zhang X H, Xue J Q, Xu S T. Identification of ear morphology genes in maize (L.) using selective sweeps and association mapping., 2020, 11: 747.
[11] Tian H L, Wang F G, Zhao J R, Yi H M, Wang L, Wang R, Yang Y, Song W. Development of maizeSNP3072, a high-throughput compatible SNP array, for DNA fingerprinting identification of Chinese maize varieties., 2015, 35: 136.
[12] Zhu X M, Shao X Y, Pei Y H, Guo X M, Li J, Song X Y, Zhao M A. Genetic diversity and genome-wide association study of major ear quantitative traits using high-density SNPs in maize., 2018, 9: 966.
[13] Zhang C S, Zhou Z Q, Yong H J, Zhang X C, Hao Z F, Zhang F J, Li M S, Zhang D G, Li X H, Wang Z H, Weng J F. Analysis of the genetic architecture of maize ear and grain morphological traits by combined linkage and association mapping., 2017, 130: 1011–1029.
[14] Xue Y D, Warburton M L, Sawkins M, Zhang X H, Setter T, Xu Y B, Grudloyma P, Gethi J, Ribaut J M, Li W C, Zhang X B, Zheng Y L, Yan J B. Genome-wide association analysis for nine agronomic traits in maize under well-watered and water-stressed conditions., 2013, 126: 2587–2596.
[15] Pandis N. Linear regression., 2016, 149: 431–434.
[16] Carraro N, Forestan C, Canova S, Traas J, Varotto S.andencode two novel putative candidates for polar auxin transport and plant architecture determination of maize., 2006, 142: 254–264.
[17] Forestan C, Meda S, Varotto S. ZmPIN1-mediated auxin transport is related to cellular differentiation during maize embryogenesis and endosperm development., 2010, 152: 1373–1390.
[18] 马娟, 曹言勇, 李会勇. 玉米穗轴粗全基因组关联分析. 作物学报, 2021, 47: 1228–1238.
Ma J, Cao Y Y, Li H Y. Genome-wide association study of ear cob diameter in maize., 2021, 47: 1228–1238 (in Chinese with English abstract).
[19] 殷芳冰, 王成, 龙艳, 董振营, 万向元. 玉米雌穗性状遗传分析与形成机制. 中国生物工程杂志, 2021, 41(12): 30–46.
Yin F B, Wang C, Long Y, Dong Z Y, Wan X Y. Progress on dissecting genetic architecture and formation mechanism of maize ear traits., 2021, 41(12): 30–46 (in Chinese with English abstract).
[20] Lu M, Xie C X, Li X H, Hao Z F, Li M S, Weng J F, Zhang D G, Bai L, Zhang S H. Mapping of quantitative trait loci for kernel row number in maize across seven environments., 2011, 28: 143–152.
[21] Karen S P, Lopes S C J, Pereira S A, Augusto F G A. QTL mapping for yield components in a tropical maize population using microsatellite markers., 2008, 145: 194–203.
[22] Tian B H, Wang J H, Wang G Y. Confirmation of a major QTL on chromosome 10 for maize kernel row number in different environments., 2014, 133: 184–188.
[23] Liu L, Du Y F, Huo D A, Wang M, Shen X M, Yue B, Qiu F Z, Zheng Y L, Yan J B, Zhang Z X. Genetic architecture of maize kernel row number and whole genome prediction., 2015, 128: 2243–2254.
[24] Ma X Q, Tang J H, Teng W T, Yan J B, Meng Y J, Li J S. Epistatic interaction is an important genetic basis of grain yield and its components in maize., 2007, 20: 41–51.
[25] Yang C, Liu J, Rong T Z. Detection of quantitative trait loci for ear row number in F2populations of maize., 2015, 14: 14229–14238.
[26] Yan J B, Tang H, Huang Y Q, Zheng Y L, Li J S. Quantitative trait loci mapping and epistatic analysis for grain yield and yield components using molecular markers with an elite maize hybrid., 2006, 149: 121–131.
[27] Brown P J, Upadyayula N, Mahone G S, Tian F, Bradbury P J, Myles S, Holland J B, Flint-Garcia S, McMullen M D, Buckler E S, Rocheford T R. Distinct genetic architectures for male and female inflorescence traits of maize., 2011, 7: e1002383.
[28] Huo D A, Ning Q, Shen X M, Liu L, Zhang Z X. QTL mapping of kernel number-related traits and validation of one major QTL for ear length in maize., 2016, 11: e0155506.
[29] Yang N, Lu Y L, Yang X H, Huang J, Zhou Y, Ali F, Wen W W, Liu J, Li J S, Yan J B. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel., 2014, 10: e1004573.
[30] Gallavotti A, Barazesh S, Malcomber S, Hall D, Jackson D, Schmidt R J, McSteen P.encodes a monocot-specific-like gene required for vegetative and reproductive development in maize., 2008, 105: 15196–15201.
[31] Phillips K A, Skirpan A L, Liu X, Christensen A, Slewinski T L, Hudson C, Barazesh S, Cohen J D, Malcomber S, McSteen P.encodes a grass-specific tryptophan aminotransferase required for vegetative and reproductive development in maize., 2011, 23: 550–566.
[32] Barazesh S, McSteen P.functions in organogenesis during vegetative and inflorescence development in maize., 2008, 179: 389–401.
[33] McSteen P, Hake S.regulates axillary meristem development in the maize inflorescence., 2001, 128: 2881–2891.
[34] McSteen P, Malcomber S, Skirpan A, Lunde C, Wu X T, Kellogg E, Hake S.encodes a co-ortholog of theserine/threonine kinase and is required for organogenesis during inflorescence and vegetative development in maize., 2007, 144: 1000–1011.
[35] Kato H, Nishihama R, Weijers D, Kohchi T. Evolution of nuclear auxin signaling: lessons from genetic studies with basal land plants., 2018, 69: 291–301.
[36] Gallavotti A, Zhao Q, Kyozuka J, Meeley R B, Ritter M K, Doebley J F, Pè M E, Schmidt R J. The role ofin the architecture of maize., 2004, 432: 630–635.
[37] Sigmon B, Vollbrecht E. Evidence of selection at thelocus during maize domestication., 2010, 19: 1296–1311.
[38] 肖朝文, 傅永福. AT-hook蛋白的研究进展. 中国农业科技导报, 2009, 11(5): 12–16.
Xiao C W, Fu Y F. Research progress in AT-hook proteins., 2009, 11(5): 12–16 (in Chinese with English abstract).
[39] Gallavotti A, Malcomber S, Gaines C, Stanfield S, Whipple C, Kellogg E, Schmidt R J. BARREN STALK FASTIGIATE1 is an AT-hook protein required for the formation of maize ears., 2011, 23: 1756–1771.
[40] Jia H T, Li M F, Li W Y, Liu L, Jian Y N, Yang Z X, Shen X M, Ning Q, Du Y F, Zhao R, Jackson D, Yang X H, Zhang Z X. A serine/threonine protein kinase encoding generegulates maize grain yield., 2020, 11: 988.
Genome-wide association study and candidate genes predication of yield related ear traits in maize
YIN Fang-Bing1, LI Ya-Nan1, BAO Jian-Xi1, MA Ya-Jie1, QIN Wen-Xuan1, WANG Rui-Pu1, LONG Yan1,2, LI Jin-Ping2, DONG Zhen-Ying1,2,*, and WAN Xiang-Yuan1,2,*
1Zhongzhi International Institute of Agricultural Biosciences, Shunde Graduate School, School of Chemistry and Biological Engineering, Research Center of Biology and Agriculture, University of Science and Technology Beijing (USTB), Beijing 100083, China;2Beijing Engineering Laboratory of Main Crop Bio-Tech Breeding, Beijing International Science and Technology Cooperation Base of Bio-Tech Breeding, Beijing Solidwill Sci-Tech Co. Ltd., Beijing 100192, China
Ear traits directly affect the final yield of maize, and the analysis of its genetic mechanisms can provide useful guidance for yield enhancement in maize. In this study, 733 maize inbred lines were planted in randomized block designs under two environments, and three yield-related traits, kernel row number (KRN), ear length (EL), and ear diameter (ED), were investigated. Genotyping was performed using MaizeSNP3072 chip and FarmCPU (fixed and random model circulating probability unification) model was used to conduct genome-wide association study (GWAS). 16, 13, and 24 single nucleotide polymorphism (SNP) loci significantly associated with the three traits were identified, and the values of phenotypic variation explained (PVE) for single locus were 0.01%–7.08%, 0.01%–5.34%, and 0.07%–4.34%, respectively. Further, six, two, and five high confidence (HC) SNPs that were repeatedly detected in multiple environments for KRN, EL, and ED were retrieved, among which two SNPs were simultaneously associated with KRN and ED traits, and one KRN HC-SNP and three ED HC-SNPs were firstly reported in this study. By searching 200 kb regions around the 11 HC-SNPs loci, 33 important candidate genes were identified, including a knowngeneregulating ear developmentauxin polar transport located in the confidence interval of chromosome 9 SNP marker PZE-109003046. Other candidate genes encoded transcription factors, hormone (such as auxin, gibberellin, and ethylene) pathway related proteins, DNA methylation, and protein phosphorylation related proteins, which might regulate ear traits by different mechanisms. The 11 HC-SNPs and 33 important candidate genes detected in this study can provide valuable information for further cloning of functional genes and reveal the molecular regulatory mechanisms and marker-assisted selection for ear trait in maize.
maize; yield related ear traits; genome-wide association study (GWAS); candidate gene
10.3724/SP.J.1006.2023.23021
本研究由国家重点研发计划项目“农业生物种质资源挖掘与创新利用”重点专项( 2021YFD1200700)资助。
This study was supported by the National Key Research and Development Program of China (2021YFD1200700).
董振营, E-mail: zydong@ustb.edu.cn; 万向元, E-mail: wanxiangyuan@ustb.edu.cn
E-mail: yinfangbing186@163.com
2022-02-27;
2022-05-05;
2022-05-26.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20220525.1950.004.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/)
