分散固相萃取法与固相萃取法在气相色谱质谱联用法测定多组分农残中的比较*
2022-11-18张月辉刘家阳李彦博
闫 玉,宋 姝,袁 宁,张月辉,刘家阳,赵 飞,于 晨,田 甜,李彦博
(辽宁省检验检测认证中心,辽宁 沈阳 110000)
现代农药技术为满足农作物多种病害虫害的防治要求,实现少次喷洒多种预防的效果,生产的农药基本为混合型农药,以多种复杂有机化合物复配而成[1-3]。因此对蔬菜水果中农药残留的检测方法要求很高,而且农药很多化合物组分结构复杂且相近,多存在同分异构现象[4],所以在食品检测中多采用气相色谱联用质谱法进行组分分析。气相色谱串联质谱仪(GC-MS/MS)检测具有灵敏度高、抗干扰能力强、选择性好等优点,正好满足农药多残留分析的需求[5-8]。
同时农药中很多化合物易分解且具有易挥发迁移等特性[9-11],而且不同蔬菜水果特性不同,在进行上机测定时,带来的干扰和影响也大不相同[12-14]。在实验中还有多次进样带来进样口、离子源的污染,导致农药中某些化合物的检测灵敏度逐渐下降,从而影响检测结果[15-17]。虽然可以通过更换衬管、割柱头以及清洗离子源等操作对仪器进行维护,但这无疑增加了检测的成本与时间。因此,对样品的净化成为提高方法耐用性的关键,所以前处理方法的选择也尤为重要,也是影响结果精确度和准确性的重要前提[18-19]。
本实验选取含有叶绿素成分较高的菠菜为作为空白基质[20],分别采用分散固相萃取(QuEChERS)和固相萃取2 种方法进行比较,通过加标回收实验,考察2 种前处理方法之间对有机磷、有机氯、拟除虫菊酯、氨基甲酸酯等多种农药化合物的回收率和准确度的差异。
1 实验部分
1.1 仪器与试剂
岛津GCMS-TQ8050 气相色谱-串联质谱仪(日本岛津公司)。
TTL-DCⅡ型氮吹仪(余姚市长江温度仪表厂)。
HeraeusMultifu.geX1R 离心机(赛默飞世尔科技有限公司)。
T25 高速匀浆机(德国ⅠKA 公司)。
乙腈、丙酮(色谱纯,Fisher 公司)。
提取盐包:4 g 硫酸镁、1 g 氯化钠、1 g 柠檬酸钠、0.5 g 柠檬酸氢二钠(迪马公司)。
净化管:900 mg 硫酸镁、150 mg 乙二胺-N-丙基硅烷化硅胶及150 mg 十八烷基硅烷键合硅胶(迪马公司)。
净化柱:石墨化碳黑-氨基复合柱。
0.22 μm 尼龙滤膜(上海安谱公司)。
实验室用水为Milli-Q 超纯水。
1.2 标准溶液配制
1.2.1 混合标准中间液(1.0 μg/mL)
准确吸取农残混合标准溶液0.50 mL 置于5 mL 容量瓶中,用丙酮溶解并稀释至刻度,配制成质量浓度为1.0 μg/mL 的农残混合标准中间液。
1.2.2 内标溶液的配制
环氧七氯标准贮备液(1.0 mg/mL):准确称取环氧七氯10.00 mg 标准品置于10 mL 容量瓶中,用丙酮溶解并稀释至刻度,配制成质量浓度为1.0 mg/mL 的环氧七氯标准贮备液。
环氧七氯标准中间液(5.0 μg/mL):准确移取上述环氧七氯标准贮备液(1.0 mg/mL)0.050 mL于10 mL容量瓶中,用丙酮稀释至刻度,配制成质量浓度为5.0 μg/mL 的环氧七氯标准中间液。
1.2.3 混合标准曲线溶液的制备
分别准确移取1.0 μg/mL 农残混合标准中间液0.05 mL、0.1 mL、0.5 mL、1.0 mL、2.0 mL 于 5 mL容量瓶,用丙酮定容至刻度,配成混合标准系列工作液,质量浓度分别为5 ng/mL、10 ng/mL、50 ng/mL、100 ng/mL、200 ng/mL。准确移取以上各浓度标准系列工作液1 mL 分别添加至经提取、净化步骤处理的5份空白样品处理吹干后的残渣中,最后加入质量浓度为5.0 μg/mL 的环氧七氯标准中间液20 μL,涡旋混合均匀,制得质量浓度分别为5 ng/mL、10 ng/mL、50 ng/mL、100 ng/mL、200 ng/mL 的系列基质标准溶液。
1.3 前处理方法
1.3.1 分散固相萃取法(QuEChERS 法)
称取5.00 g 粉碎后的试样(精确至0.01 g)于50 mL塑料离心管中,加入10 mL 乙腈、4 g 硫酸镁、1 g 氯化钠、1 g 柠檬酸钠、0.5 g 柠檬酸氢二钠及1 颗陶瓷均质子,盖上离心管盖,剧烈震荡1 min 后4 200 r/min离心5 min。吸取6 mL 上清液加到内含900 mg 硫酸镁及150 mg PSA 的15 mL 塑料离心管中。涡旋混匀1 min,4 200 r/min 离心5 min,准确吸取1 mL 上清液于10 mL 试管中,40 ℃水浴中氮气吹至近干。加入质量浓度为5 μg/mL 的环氧七氯内标溶液20 μL,加入1 mL 丙酮复溶,涡旋混匀,过微孔滤膜,用于测定。
1.3.2 固相萃取
称取5.00 g 粉碎后的试样(精确至0.01 g)于50 mL塑料离心管中,加入10 mL 水涡旋混匀,静置30 min,加入20 mL 乙腈,15 000 r/min 高速匀浆2 min,加入5 g 氯化钠剧烈振荡,5 000 r/min 离心5 min,准确吸取5 mL 上清液,40 ℃水浴旋蒸至1 mL,氮气吹至近干,待净化。用5 mL 乙腈-甲苯溶液(3+1)预洗石墨化碳黑-氨基复合柱,弃去流出液。下续150 mL 鸡心瓶,放入固定架上。将上述待净化试样用3 mL 乙腈-甲苯溶液(3+1)洗涤至固相萃取柱中,再用2 mL 乙腈-甲苯溶液(3+1)洗涤,并将洗涤液移入柱中,重复2 次。用25 mL 乙腈-甲苯溶液(3+1)淋洗固相萃取柱,收集上述所有流出液于150 mL 鸡心瓶中,40 ℃水浴中旋转浓缩至近干。加入5.0 μg/mL 环氧七氯内标溶液20 μL,加入1.0 mL 丙酮复溶,过微孔滤膜,用于测定。
1.4 仪器条件
1.4.1 色谱条件
色谱柱:SH-Rtx-1701(30 m×0.25 mm(内径)×0.25 μm(膜厚))。
柱温:40 ℃保持1 min,以40 ℃/min 的速率升温至120 ℃,保持0 min,以5 ℃/min 的速率升温至240 ℃,保持 0 min,以 12 ℃/min 的速率升温至 300 ℃,保持10 min。
进样口温度:280 ℃。
载气(高纯氦气)线速:36.1 cm/s 恒线速。
进样量:1 μL。
1.4.2 质谱条件
接口温度:280 ℃。
离子源:电子轰击源(EⅠ源)。
离子源温度:230 ℃。
扫描方式MRM。
农残化合物质谱条件如表1 所示。
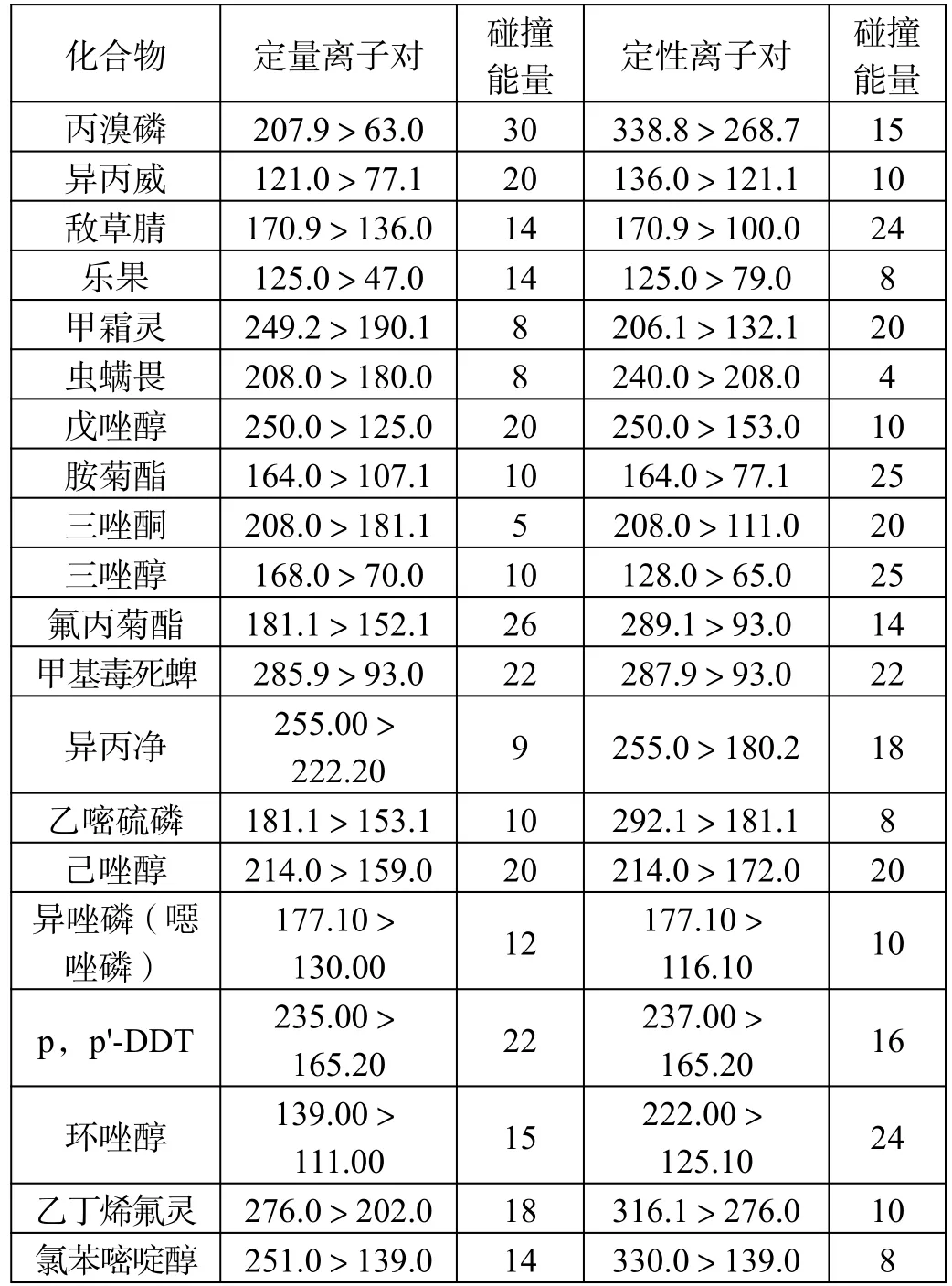
表1 农残化合物质谱条件
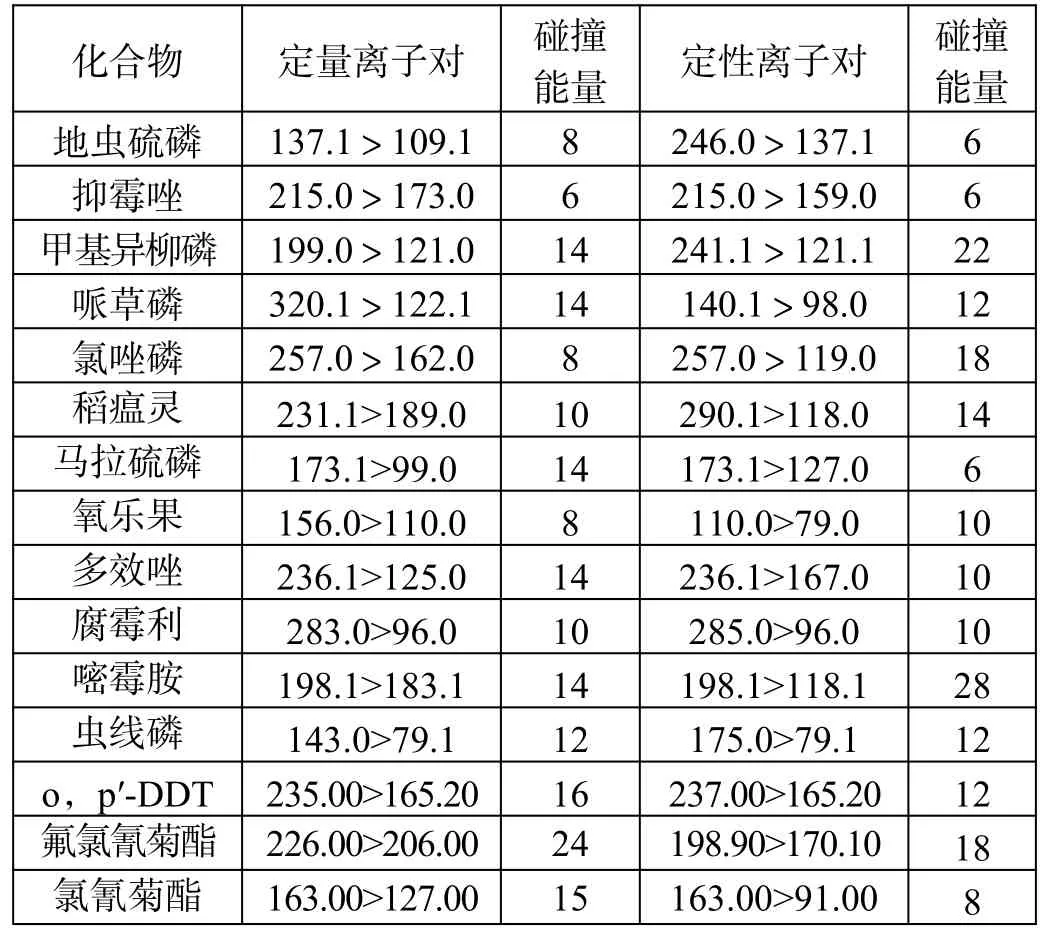
表1(续)
2 结果与讨论
2.1 分散固相萃取法
标准曲线、线性相关性、加标回收率及RSD 值如表2 所示。
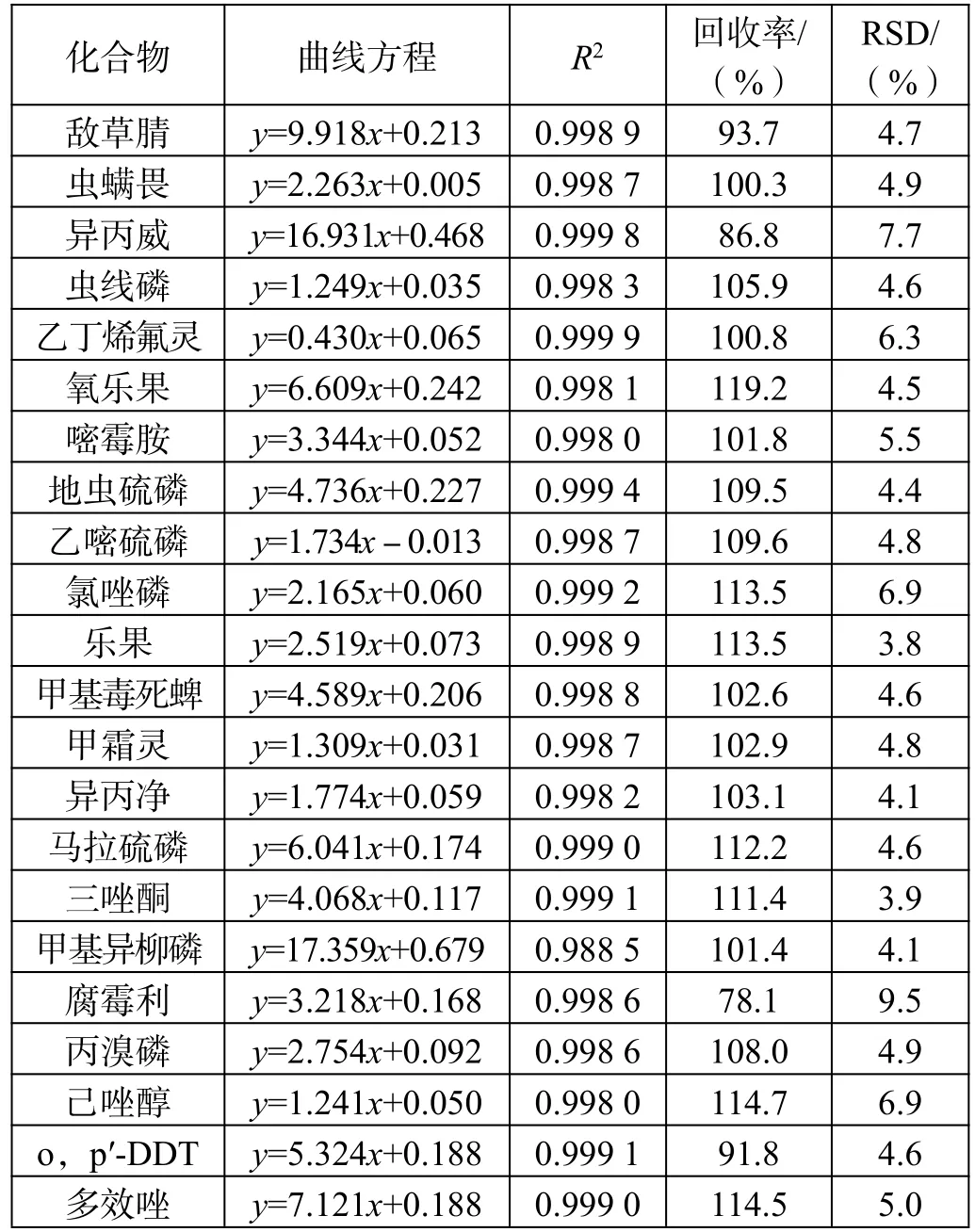
表2 分散固相萃取法所得曲线方程、线性相关性、加标回收率及RSD 值
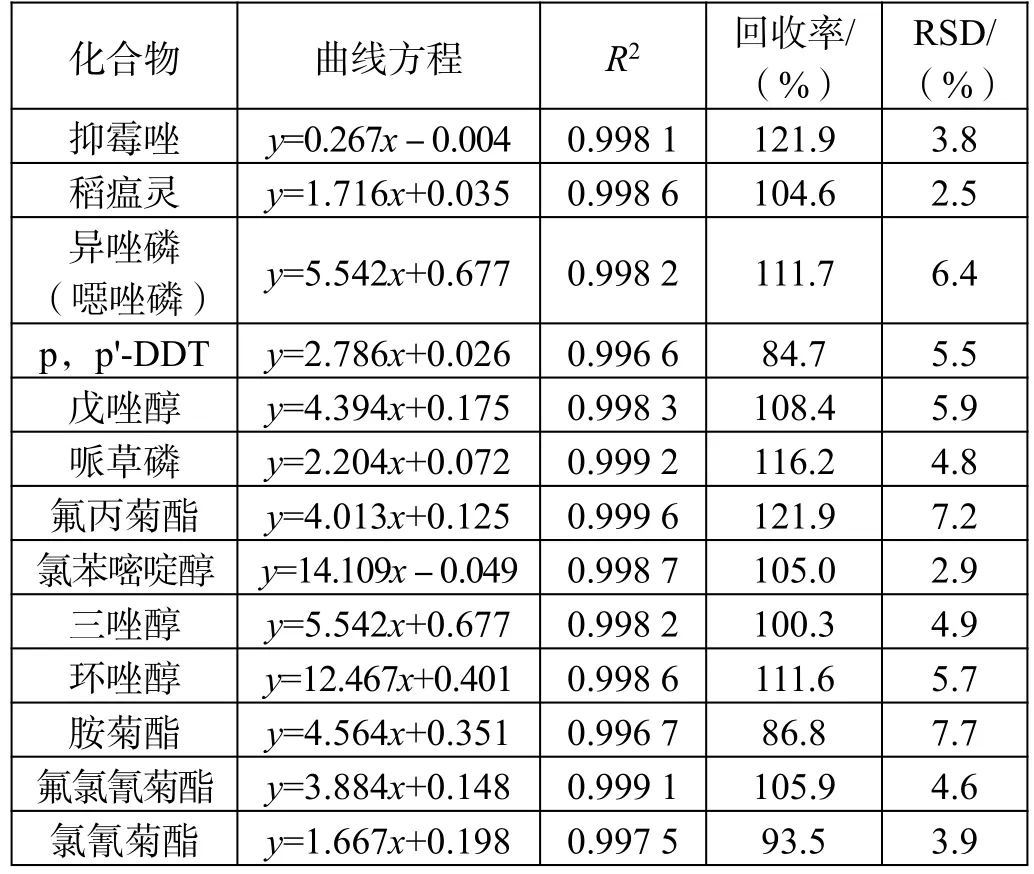
表2(续)
加标回收实验方法:准确称取5.000 g 样品6 份,分别加入农残混合标准中间液(1.0 μg/mL)0.10 mL,提取、净化过程同样品处理,进样,测定回收率,计算 RSD 值。
2.2 固相萃取法
标准曲线、线性相关性、加标回收率及RSD 值如表3 所示。
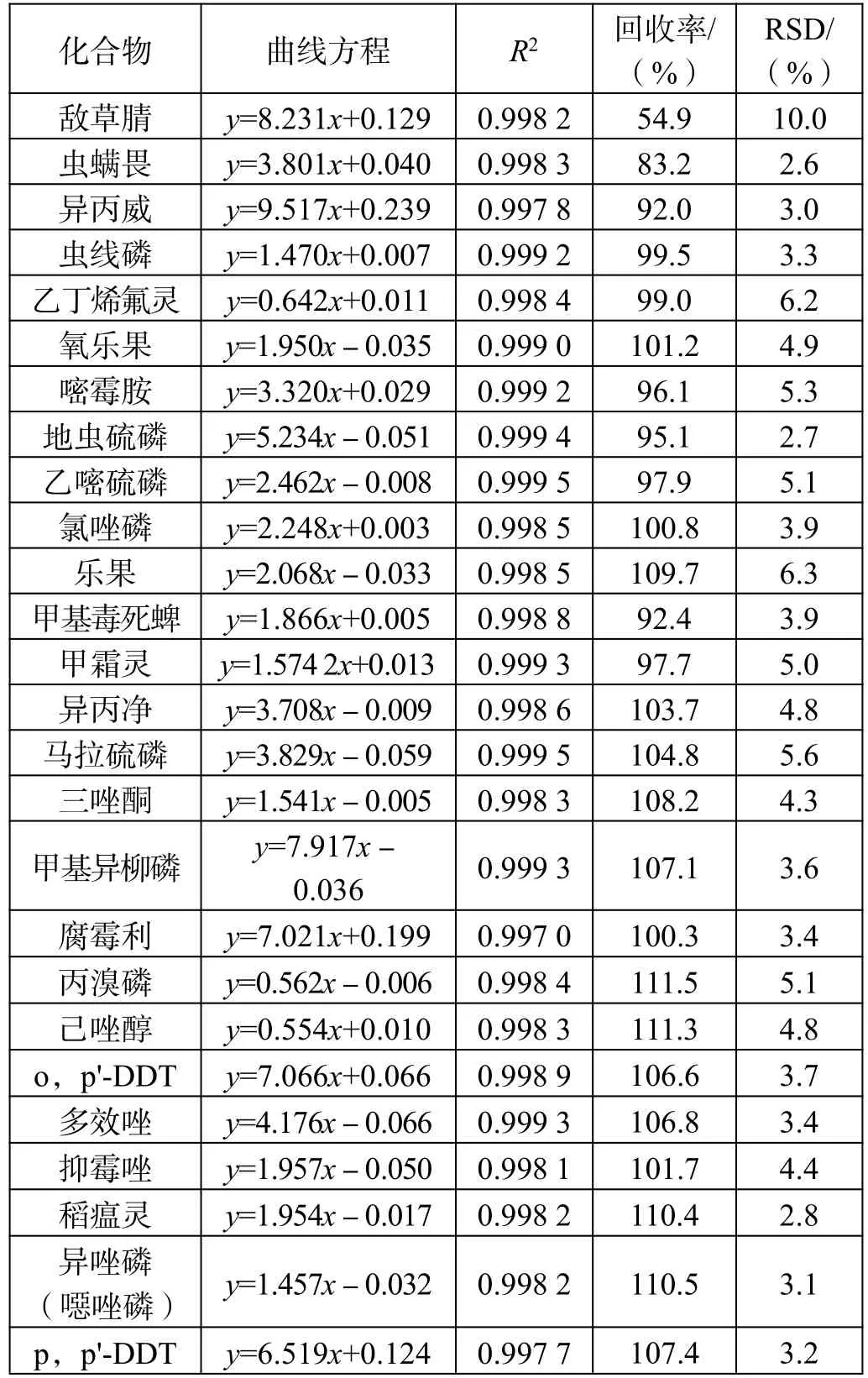
表3 固相萃取法所得曲线方程、线性相关性、加标回收率及RSD 值
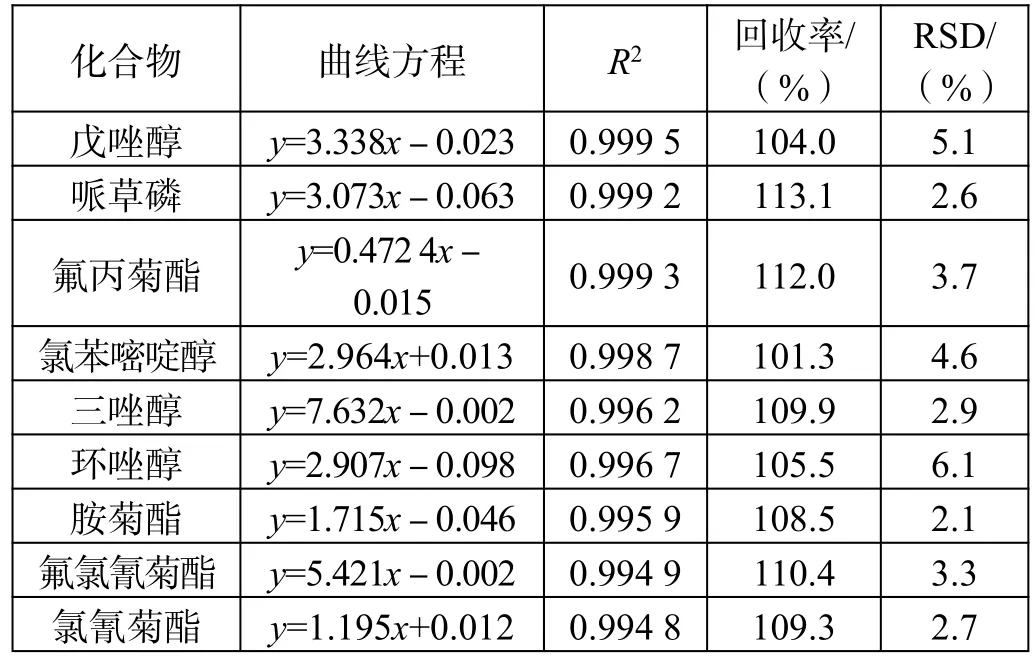
表3(续)
加标回收实验方法同2.1。
2.3 分析讨论
从结果可以看出该仪器条件下,标准曲线线性相关性均满足一般实验对结果的要求,35 种农药成分的线性相关性均在0.995 以上。
从RSD 值来看,分散固相萃取法和固相萃取法的平均标准偏差均小于10,数据重现性较好,2 种前处理方法均可满足实验要求。
通过对比每个组分的加标回收率可以发现,部分组分农药采用分散固相萃取前处理方法的加标回收率优于采用固相萃取。这是由于固相萃取法中使用的氨基炭黑复合柱中吸附结构致密紧实,吸附性强,而很多农药的分子结构多为长链性和平面片层结构,被吸附后不易洗脱,造成回收率较低;虽然分散固相萃取组分也含有吸附能力的物质,但由于结构松散,农药组分回溶较为容易,所以回收率反而较高。
3 结论
通过对比这2 组实验结果可以看出,分散固相萃取法可以很好地实现多组分农残的检测,部分组分的回收率优于固相萃取法。分散固相萃取法相较于固相萃取法减少了净化和洗脱环节,有效节省了有机试剂的使用量,缩短了前处理时间,且分散固相萃取管的价格远低于固相萃取柱,在保证实验快捷、高效、结果准确性的前提下,还有效降低了实验成本。
