甘草素固体分散体的制备及体内药动学研究
2022-11-16张军唐粤鹏陈剑大理大学药学院云南大理67000上海交通大学药学院上海0040
张军,唐粤鹏,陈剑*(.大理大学 药学院,云南 大理 67000;.上海交通大学 药学院,上海 0040)
甘草素(liquiritigenin,LQ)是一种从豆科植物中提取的二氢黄酮单体化合物,在自然界中主要以糖苷苷元形式存在。目前发现甘草素具有抗肿瘤[1]、抗炎[2]、抗抑郁[3]、抗过敏[4]、保护成骨细胞[5]等作用。甘草素为生物药剂学分类系统(BCS)中Ⅱ类化合物,其溶解度低导致口服生物利用度很低,限制了该化合物的进一步临床应用。
为解决这一问题,研究者将其溶于pH 4.0~6.0,含量为40%的丙二醇溶液中制备为注射液[6]。在口服制剂方面,该团队开发了甘草素的磷脂复合物和亚微米乳液,将其口服生物利用度提高至2.39 倍和5.95 倍[7-8]。
制备固体分散体是增加难溶性药物的溶解度,提高其口服生物利用度的常见方法。该技术简单、高效,通过将药物转化为无定型状态,增强药物的溶出。截至2020年,FDA 已经批准26 种通过该技术上市的药物[9]。其中,多数制剂使用聚乙烯吡咯烷酮K30(PVP K30)作为载体。PVP 是最常研究的亲水性聚合物载体之一[10],具有良好的稳定性和溶解性,可防止无定形药物结晶。PVPK30广泛地应用于各种难溶性药物的固体分散体中,如尼索地平[11],吲哚美辛[12],姜黄素[13]等。
目前尚未见甘草素口服固体制剂的报道,本研究拟采用溶剂蒸发法,以PVP K30 为载体,介孔二氧化硅为吸收剂[14-15],制备了一种能够提高口服生物利用度的口服固体制剂甘草素固体分散体。以差示扫描量热法(DSC)、X 射线衍射分析(XRD)和红外光谱(IR)对固体分散体进行表征,并通过体外溶出和体内药代动力学实验,考察该制剂的溶出行为和口服生物利用度。
1 材料
1.1 仪器
RC806 型溶出试验仪(天津市天大天发科技有限公司);STA449F3 型同步热分析仪(德国耐驰公司);D8DaVinci 型多功能X 射线衍射仪(德国布鲁克公司);BZF-30 型真空干燥箱(上海博迅医疗生物仪器股份有限公司);1290 型超高效液相色谱仪(安捷伦科技有限公司);Nicolet 6700 型红外光谱仪(美国赛默飞世尔科技公司);5415 R 型高速离心机(德国艾本德股份公司)。
1.2 试药
甘草素原料药(HPLC ≥98%,四川维克奇生物科技有限公司);聚乙烯吡咯烷酮K30(批号:010420001,重庆斯泰克瑞登梅尔材料技术有限公司);介孔二氧化硅(批号:1000356636,德国Syloid 公司);氢氯噻嗪(HPLC ≥98%,上海易恩化学技术有限公司);无水乙醇(上海麦克林生化公司);乙腈为色谱级。
1.3 动物
SPF 级健康SD 大鼠,雄性,体质量200~230 g[上海吉辉实验动物饲养有限公司,许可证号:SCXK(沪)2017-0012]。所有涉及大鼠的操作均按照《实验动物护理与使用指南》及相关中国法律法规的要求进行,并经上海交通大学机构动物护理与使用委员会批准(批准号A2021052)。
2 方法
2.1 甘草素的分析方法
2.1.1 色谱条件 色谱柱:Agilent Eclipse Plus RRHD C18(2.1 mm×100 mm,1.8 μm); 流动相:0.1%乙酸溶液-乙腈(60∶40);流速:0.1 mL·min-1;检测波长:278 nm;柱温:30℃;进样量:5.0 μL。
出席本次展览开幕式的嘉宾有:浙江省文联书记处书记张均林,浙江省文联党组成员、书记处书记徐晓,浙江省直住房公积金管理中心主任应金龙,浙江省美术家协会副主席、浙江画院院长孙永,嘉兴日报原社长张扣林,浙江省美术家协会副主席、浙江画院副院长池沙鸿,浙江画院党支部书记、山水画工作室主任茹峰,浙江画院专职画师余昌梅,北京华夏十德文化研究院副院长徐茹,浙江省艺术品鉴赏研究会会长管建平,海宁天桐家文化传媒有限公司董事长陈国兴等;兰溪市领导嘉宾有:中共兰溪市委书记朱瑞俊,兰溪市委常委、宣传部长翁柯卫,兰溪市人大副主任胡向东,兰溪市政协副主席陈兴兵。开幕式由兰溪市副市长吴丽娅主持。
2.1.2 线性关系 精密称取100 mg 甘草素于100 mL 量瓶,加入甲醇完全溶解并定容,得到1 mg·mL-1的对照品储备液;用流动相依次稀释,得到1、5、10、25、50、100、150 μg·mL-1的对照品溶液,进样测定。以其质量浓度(X)对甘草素峰面积(Y)进行回归,得到回归方程Y=153.4X+59.01(R2=0.9998),甘草素在1~150 μg·mL-1与峰面积线性关系良好。甘草素理论塔板数为2455。
2.1.3 方法学验证 取上述1、25、150 μg·mL-1的甘草素对照品溶液,平行进样测定6 次,测定RSD分别为0.19%、0.19%、0.30%,表明仪器精密度良好。取“2.2”项下10 mg 甘草素固体分散体于100 mL 量瓶,加入流动相超声溶解、定容,使用0.22 μm 滤膜过滤,得到供试品溶液。取续滤液,于0、2、4、6、12、24 h 进样测定,测得RSD为0.61%,表明溶液在24 h 内稳定性良好。取10 mg甘草素固体分散体6 份,制备成供试品溶液,进样测定,测得RSD为0.57%,表明该方法重复性良好。取10 mg 甘草素固体分散体于100 mL 量瓶,分别加入“2.1.2”项下对照品储备液2、4、6 mL,加入流动相超声溶解并定容,使用0.22 μm 滤膜过滤,得到9 份供试品溶液,进样测定,测得低、中、高浓度甘草素的平均加样回收率为99.72%、100.75%、101.50%,RSD为1.6%、2.0%、0.73%。
2.2 甘草素固体分散体的制备
按LQ∶PVP K30 为1∶1、1∶4、1∶6、1∶8、1∶12 的比例称取原料于烧杯中,加入适量的无水乙醇密封,于室温下,磁力搅拌1 h;加入50%(w/w)的介孔二氧化硅作为吸收剂,继续搅拌2 h 后,于40℃旋转蒸发仪除去大部分溶剂,残余溶剂于40℃真空干燥过夜。将所得固态混合物转移至研钵中,粉碎,过60目筛,即得甘草素固体分散体。
2.3 物理混合物的制备
称取100 mg 甘草素原料药、800 mg PVP K30和450 mg 介孔二氧化硅,采用等量倍增法混合均匀,过60 目筛,置于干燥器中保存,备用。
2.4 含量测定
精密称取适量甘草素固体分散体分散于乙醇中,水浴超声10 min 后,用乙醇定容至10 mL,摇晃均匀后,过0.22 μm 滤膜,用流动相稀释适当倍数,进样测定。
2.5 体外溶出研究
参照《中国药典》(2020年版)中的“溶出度与释放度测定法”第三法小杯法,测定甘草素固体分散体的体外溶出度。转速为50 r·min-1,温度为(37±0.5)℃,溶出介质为含0.25% Tween 80的0.1 mol·L-1HCl 溶液,介质体积为150 mL。取甘草素和各处方量制备得到的固体分散体(相当于甘草素20 mg),置于溶出杯中,分别于5、15、30、45、60、90、120 min 取样,每次每缸取样2 mL(同时补充等量同温介质),经0.22 µm 水相微孔滤膜过滤,取续滤液1.0 mL,进样测定,计算溶出度,绘制溶出曲线,并采用非模型依赖法——f2法对溶出曲线进行评价[16]。
2.6 DSC 分析
取适量甘草素原料药、PVP K30、介孔二氧化硅、物理混合物、甘草素固体分散体,放于氧化铝坩埚内,在纯氮气下升温,温度范围为40~250℃,速度10 ℃·min-1,进行DSC 分析。
2.7 XRD 分析
为测定药物的结晶特征,取适量甘草素原料药、PVP K30、介孔二氧化硅、物理混合物、甘草素固体分散体粉末,进行XRD 分析。测定条件为X 线光源 Cu 靶,陶瓷X 光管,电压≤40 kV,电流≤40 mA;扫描速度0.2 S;扫描范围2θ5°~60°。
2.8 IR 分析
2.9 体内药代动力学
2.9.1 给药方案及血浆样品的采集 取健康SD雄性大鼠6 只,随机分为实验组和对照组,每组3 只大鼠,给药前禁食不少于12 h,自由饮水,进行单次、单剂量给药实验。对照组灌胃给予相当于20 mg·kg-1的纯甘草素混悬液,实验组灌胃给予相当于20 mg·kg-1的甘草素固体分散体混悬液。给药后,分别于5、15、30、45、60 min 及1.5、2、3 h,眼眶取血0.5 mL,置于EDTA 抗凝管中,于4 ℃、4000 r·min-1离心5 min,取上层血清,于-20℃冰箱中冷冻备用。
2.9.2 血浆样品的处理 取血浆样品100 μL,置于1.5 mL 离心管中,加入内标溶液(氢氯噻嗪,37.7 μg·mL-1)40 μL,涡旋混合30 s。加入1 mL乙醚,涡旋混合3 min,4℃、1 610 000 r·min-1转速离心5 min 后,转移上层溶液,于常温真空干燥箱40℃挥干。用30 μL 流动相复溶,涡旋5 min后,10 000 g 离心5 min,取上清液,待测。
2.9.3 血药浓度分析方法
① 线性关系考察:配制质量浓度为10 μg·mL-1的甘草素溶液,使用甲醇稀释至0.02、0.08、0.15、0.3、0.6、1.2 μg·mL-1,体积为100 μL。氮气吹干后加入100 μL 空白血浆,按照“2.9.2”项下方法对样品进行处理,进样测定。以甘草素质量浓度为横坐标(X),甘草素与内标物的峰面积比值(Y)为纵坐标,得到线性回归方程:Y=1.595×10-4X+0.066 25,R2=0.9997,线性范围为20~1200 ng·mL-1。
② 方法学考察:制备质量浓度0.02、0.3、1.2 μg·mL-1的血浆对照品溶液,进样测定6 次,测定甘草素与内标峰面积比值RSD分别为7.1%、2.3%、1.3%,表明仪器精密度良好。取一份血浆样品,于0、2、4、6、12、24 h 冷冻溶解后,按照“2.9.2”项下方法进行处理,进样检测,测得RSD为9.0%,表明血浆样品在24 h 内具有良好的稳定性。取空白血浆,配制20、300、1200 ng·mL-1质量浓度的血浆对照品溶液,按照“2.9.2”项下方法进行处理,进样测定,计算加样回收率分别为83.80%、88.85%、94.83%,RSD分别为3.8%、1.6%、0.42%。取100 μg·mL-1的储备液,经过流动相逐级稀释,以信噪比3、10 为检测限、定量限,测得两者分别为5 ng·mL-1、10 ng·mL-1。
③ 数据处理:统计分析采用DAS 2.0 软件计算药代动力学参数。所有数值均以均值±标准差(SD)表示。采用t检验进行统计分析。P<0.01为差异有统计学意义。
3 结果
3.1 体外溶出实验结果
如图1所示,物理混合物和固体分散体均可以提高甘草素的溶出效率,而固体分散体相较于物理混合物更加显著。原料药、物理混合物在120 min 内的累积溶出率分别为(30.10±1.95)%、(45.33±4.93)%;而在制备为固体分散体后,药物的溶出率显著增强,并且随着PVP K30 用量的增加,药物的溶出速度与累积溶出率逐渐提高。当LQ∶PVP K30 为1∶8、1∶12 时,前45 min 内的累积溶出率超过80%,前120 min 内累积溶出率达到95%。对几个处方在前45 min 的溶出曲线进行了相似因子f2的计算。当f2在50~100 内时,认为两条溶出曲线相似[16]。通过计算发现,LQ∶PVP K30 比例为1∶6 和1∶8 的两个处方溶出曲线之间的f2是49.79,而1∶8 和1∶12 之间的f2是55.81。因此,可以认为当两者比例达到1∶8 之后,溶出速度不再有显著的增长,后期实验中选择LQ∶PVP K30比例为1∶8作为制剂处方。

图1 甘草素各制剂处方溶出曲线(n=3)Fig 1 In vitro release curve of liquiritigenin and its preparation(n=3)
3.2 DSC 分析结果
在DSC 结果中,甘草素原料药在212.19 ℃处有一个明显的尖峰,此为甘草素的熔点峰;PVP K30 和介孔二氧化硅在40~250 ℃没有明显的吸收峰;甘草素固体分散体和原料药相比,在212.19 ℃处的吸收峰消失,表明甘草素在固体分散体中是以无定型的状态分布(见图2)。
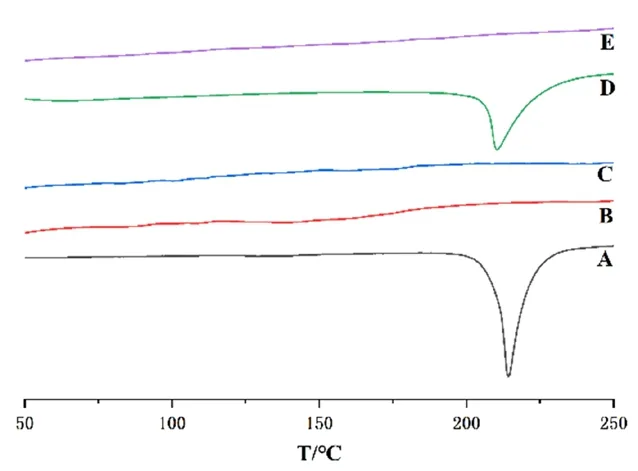
图2 甘草素原料药(A)、PVP K30(B)、介孔二氧化硅(C)、物理混合物(D)、固体分散体(E)的DSC 图谱Fig 2 DSC curves of liquiritigenin(A),PVP K30(B),SiO2(C),physical mixture(D)and LQ-SD(E)
3.3 XRD 分析结果
甘草素原料药在2θ为10.8°和16.0°附近有较强的吸收峰,在16°~29.3°附近存在一系小峰,表明原料药中甘草素以结晶状态存在;PVP K30 及介孔二氧化硅并未出现明显的吸收峰。物理混合物中,甘草素的吸收峰仍然存在,表明简单的物理混合过程并没有改变甘草素的晶型存在状态。在甘草素固体分散体中,甘草素的特征吸收峰完全消失,表明经过制剂加工,甘草素失去了原有的晶型状态,以无定型的形式分散其中(见图3)。
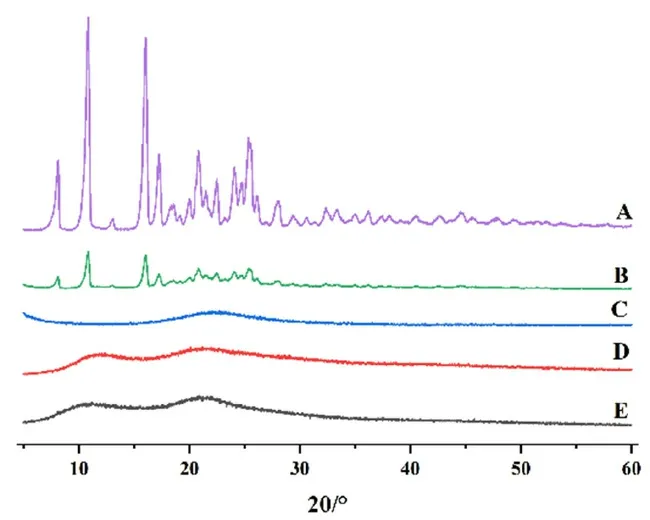
图3 甘草素原料药(A)、物理混合物(B)、PVP K30(C)、介孔二氧化硅(D)、固体分散体(E)的XRD 图谱Fig 3 XRD curves of liquiritigenin(A),physical mixture(B),PVP K30(C),SiO2(D),and LQ-SD(E)
3.4 IR 分析结果
在甘草素红外谱图中(见图4),3414~3000 cm-1处的强峰以及1600~1000 cm-1内的强峰,是由甘草素分子中O-H、C-O 以及苯环上C=C 伸缩振动引起的,验证了分子中苯环和酚羟基的存在;在1650 cm-1处有一个较强吸收峰,为C=O 的伸缩振动引起,并且其与苯环相邻,由于共轭效应的存在,使羰基的吸收向低波数移动。在PVPK30 的谱图中,3423 cm-1和1647 cm-1处的吸收峰是由是由O-H、C=O 的伸缩振动引起的[17]。对比物理混合物和固体分散体的谱图,固体分散体的图谱中C=O 的吸收峰从1650 cm-1移至1661 cm-1,表明甘草素与载体之间发生了相互作用力[7,18-19];两者中仍然可以看到甘草素的特征吸收峰,表明甘草素在制备处理过程中未受到影响。
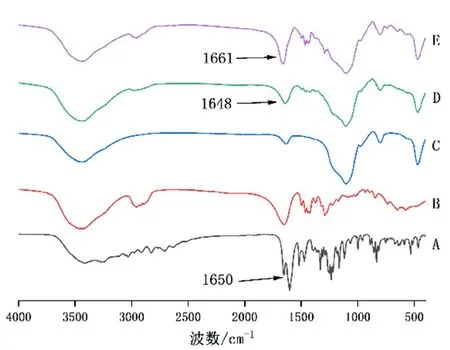
图4 甘草素原料药(A)、PVP K30(B)、介孔二氧化硅(C)、物理混合物(D)、固体分散体(E)的红外图谱Fig 4 IR of liquiritigenin(A),PVP K30(B),SiO2(C),physical mixture(D)and LQ-SD(E)
3.5 体内药代动力学
血药浓度-时间曲线如图5所示,DAS 软件处理得到的药代动力学参数如表1所示。与甘草素原料药相比,口服固体分散体后,最大血药浓度提高至6.14 倍,相对生物利用度增加至7.08倍,表明该制剂可以促进甘草素的吸收。

表1 甘草素原料药和固体分散体的主要药代动力学参数(n=3)Tab 1 Main pharmacokinetic parameters for LQ and LQ-SD (n=3)
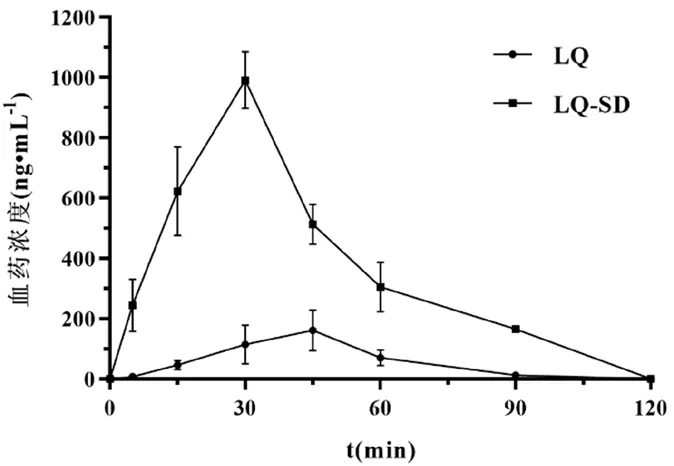
图5 甘草素血药浓度-时间曲线(n=3)Fig 5 Plasma concentration-time curves for LQ and LQ-SD(n=3)
4 讨论
固体分散体技术在药学领域的应用已经十分广泛。很多研究表明,将难溶性药物制备成固体分散体,能够显著提高药物在生物体内的生物利用度。甘草素作为BCS 中的Ⅱ类化合物,也具有溶解性差的显著特点。由于目前对其固体分散体的剂型研究较少,本研究采用PVP K30 为载体,制备出甘草素固体分散体制剂。
在研究过程中,考察了PVP K30 的用量对甘草素溶出行为的影响,药物的溶解随着载体含量的增加而增加,高比例的PVP K30 在固体分散体中的溶解速率显著提高,原因可能是PVP K30 是一种亲水性聚合物,能够与药物作用形成氢键,并且对介质的表面张力有降低作用,导致疏水药物表面的润湿,扩大了溶解的表面积,从而提高固体分散体样品的溶出速率[10]。在制备过程中,加入介孔二氧化硅作为吸收剂,可以有效地抑制无定型的药物析出结晶[20],二氧化硅表面的硅烷醇基团还可以通过提高药物颗粒的润湿性而加快溶解速度[21]。
经过各项试验证明,甘草素能够以无定型状态分布于固体分散体中,体外溶出可达到95%,有效提高了甘草素的溶出度。之前有文献报道将甘草素制成磷脂复合物,相对生物利用度提高至239%[7]。本研究大鼠体内药代动力学的结果显示,甘草素固体分散体相对生物利用度增加至7.08 倍,有更好的改善效果。
综上所述,本研究制备出的甘草素固体分散体溶解性好、相对生物利用度高,为提高甘草素的口服生物利用度提供了新的思路。
