蛋白质结构预测的方法探究
2022-11-03王芳李洪进李虎阳
王芳,李洪进,李虎阳
(遵义医科大学 医学信息工程学院,贵州 遵义 563000)
0 引言
蛋白质旧称“朊”,是一种复杂的有机化合物,是生命的物质基础,约占人体重量的16%~20%,是人体一切细胞、组织的重要组成部分。
氨基酸是构成蛋白质的基本单位,在脱水缩合的方式下形成连接两个氨基酸分子的肽键,进而连接形成肽链,如图1所示。氨基酸按照不同的比例组合成种类繁多的蛋白质,且不同蛋白质的性质和功能各不相同。
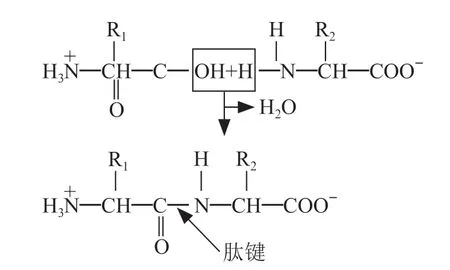
图1 氨基酸脱水后形成肽键
蛋白质结构的不同决定了蛋白质之间的功能差异。一般而言,蛋白质具有一到四级结构,如图2所示。
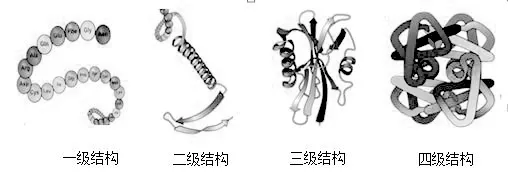
图2 蛋白质的结构示意图
(1)一级结构(primary structure)。该结构属于一维空间结构,是指氨基酸的残基在肽链中的排列顺序为线性氨基酸序列,该定义仅适用于简单的蛋白质,其形态千变万化。(2)二级结构(secondary structure)。常用来描述肽链按一定的规律卷曲或折叠的特定空间结构,是一种稳定且有限的结构,比如α-螺旋结构和β-折叠结构。(3)三级结构(tertiary structure)。是指蛋白质分子中的肽链在二级结构的基础上发展形成更为复杂的三维结构,是蛋白质的高级结构,通过二级结构预测三级结构是目前研究的重点课题。(4)四级结构(quaternary structure)。是指具有独立三级结构的多肽链通过相互作用组合而成的聚集体结构,由于其可分离,所以属于一种不稳定的结构,是蛋白质最高级的结构。
迄今已有约一千种蛋白质的一级结构被研究确定,如胰岛素、胰核糖核酸酶、胰蛋白酶等。由于蛋白质只有在折叠成特定结构之后才能行使其生物学功能,如若蛋白质折叠错误,则会导致蛋白质行使错误的功能,比如阿尔兹海默症病患者的显微病理呈现的神经纤维缠结主要由高度磷酸化的微管相关的Tau 蛋白异常折叠聚集而成。
了解蛋白质的结构有助于我们更好地认识蛋白质的功能、功能机制和执行方式,充分发掘其生物信息,因此正确预测蛋白质结构对于生物学、医药学等领域的研究发挥着至关重要的作用。
蛋白质的折叠并非全是自发折叠,其还会受很多外在因素(作用力)的影响。虽然对蛋白质折叠机理的研究是生命科学领域的前沿课题,吸引了很多物理学家和生物学家的广泛关注,但由于蛋白质结构的复杂性,我们对蛋白质的折叠过程仍然知之甚少。一些理论研究和实验结果使得我们对蛋白质的折叠过程有了更加深入的了解,但仍然存在很多悬而未决的问题。
1 蛋白质结构预测方法
由于蛋白质序列数目的快速增长,随之而来的庞大数据使得传统的试验方法无法与之更新速度相匹配,所以新理论新方法的诞生,为研究蛋白质结构和功能之间的关系提供了广阔的平台。随着计算机技术的高速发展,对蛋白质结构的预测与计算机科学之间也产生了密不可分的联系。近年来,基于计算机理论来预测蛋白质二级结构的方法迅猛发展,比如模糊聚类分析、贝叶斯分类方法、马尔科夫链、支持向量机(SVM)、K 近邻、人工神经网络(ANN)等,其中部分分析方法的预测精度较好。
采用PseAA方法提取蛋白质序列特征,弥补了常用方法AAC信息易丢失的弊端,并在引入近似熵、疏水性模式以及图像处理方法进行提取的基础上,张安胜等人提出一种基于深度学习的蛋白质二级结构预测方法。在蛋白质序列特征的提取中,用一个30-D 特征向量来表示一条蛋白质序列,利用近似熵、疏水模式以及图像处理方法等获取蛋白质序列的特征向量,其中近似熵用来估算蛋白质序列的复杂度,并给出了近似熵的计算方法。在对二级结构进行预测时,使用四个限制玻尔兹曼机(Restricted Boltzmann Machine,RBM)堆叠而成的深度玻尔兹曼机(DBM),该DBM 结构包括一个输入层(由一个30-D 蛋白质特征向量构成)、一个输出层(由一个4-D 向量构成,用来表示蛋白质所属类别)、4 个隐藏层。通过样本对RBM 逐层训练,在训练中采用Hinton 提出的逐层贪婪优化训练的策略(快速近似算法,CD)得到与输入的30-D蛋白质特征向量类似分布的输出作为微调(采用BP 算法)的权值。
采用k 维交叉验证法对Z498 和FC699 数据集进行了实验,结果表明这种蛋白质二级结构预测方法的预测准确率较好,在Z498 上可达92.78%,且在FC699 上的预测精度(63.4%)高于传统的预测方法(贝叶斯55.8%、K 近邻
56.9%、SVM62.5%、ANN63.1%)。
通过融合蛋白质序列的多种信息,利用多维尺度分析对位置特异性得分矩阵的自协方差转换(ACCPSSM)提取的进化信息进行降维,以减少机器学习的计算成本。利用PseAAC 方法提取蛋白质序列信息,通过二维小波降噪去除冗余,采用SVM 分类算法处理高维数据,提出一种有效提高蛋白质结构类预测准确性的新方法,该方法通过PORTER 在线服务进行二级结构信息的提取,如图3所示。

图3 基于PseAAC 方法的蛋白质结构类预测流程图
在蛋白质结构的预测中,特征的提取和分类是两个关键环节。为提高预测的准确性,在蛋白质的特征提取中采用最基础的特征提取方法——氨基酸组分法,即通过计算20 种天然氨基酸的频率来标记其位置信息,在多信息融合的基础上使融合后的信息代表性更强,预测效果更好。
预测分类使用了SVM 算法,主要是通过非线性变换完成输入的样本空间变量到高维的希尔伯特空间的转换,以获取最优线性分类面。在三个标准低相似蛋白质结构类数据集25PDB、640、1189 上总体预测准确度分别为93.1%、89.5%、90.8%,与近十年的预测方法相比,该算法的预测效果较好。
进化算法(Evolutionary Computation,EC)以达尔文的进化论为基础,是一种基于自然选择和遗传变异的全局性搜索算法,广泛应用于蛋白质结构的预测中。信息熵的概念抽象,在数学建模中用来描述事物的不确定性,也常被理解为某种特定信息出现的概率。在进化算法的基础上,谢腾宇等人提出的基于接触图残基对距离约束的蛋白质结构预测法和基于信息熵的蛋白质结构预测方法,将蛋白质结构的预测过程分为探索阶段和增强阶段,如图4所示,在一定程度上降低了蛋白质空间随氨基酸序列长度指数增加而带来的计算差异性,并创建了基于接触图的预测模型评价体系,从而反应该算法的预测能力。该方法通过残基位的二面角分布信息来构建信息熵,利用得到的归一化因子反映种群的多样性。

图4 核心算法示意图
在对PDB 数据库中30 个测试蛋白的实验结果显示,探索阶段的迭代次数高于2 000 时,该测试方法的预测精度更高,某些蛋白质的预测结构更接近于天然结构;迭代次数小于2 000 时,与传统预测方法相比效果无显著差异,该测试方法在某些蛋白质区域中的预测优势不明显。
遗传算法起源于对生物系统进行的计算机模拟研究,是计算数学中用于实现最佳化的搜索算法,且不依赖于某一具体问题,能够用于极其复杂的优化计算,是进化算法中的一种。近年来,遗传算法广泛应用于蛋白质结构的预测模拟中,且取得了较好的预测结果。杨瑶提出的基于改进遗传算法的蛋白质三维结构预测研究,采用惩罚函数和海明距离优化了蛋白质结构预测的思路和方法。该方法弥补了传统遗传算法中搜索范围逐渐变窄的弊端,协调了变异率与收敛速度的取舍等问题。通过改变交叉运算的随机性,进行独立的选择、交叉和变异运算,引入小生境技术辅助遗传算法实现真正的全局最优解。通过对PDB 数据库中11 条真实蛋白质进行实验对比,与传统的算法(禁忌搜索法和PERM 算法)相比,改进的遗传算法在运算速度和最优解精度上均较优,尤其是在长序列蛋白质结构的预测上优势明显。
2 现有蛋白质数据库
蛋白质数据库中大量已被标识的蛋白质序列、结构和功能为诸多研究者提供了更为广阔和便利的科研平台。常见的蛋白质数据库包括蛋白质序列数据库、蛋白质结构数据库和蛋白质分类数据库。
SWISS-PROT(https://beta.uniprot.org/)是一个高质量的人工注释和非冗余的蛋白质序列数据库,汇集了实验结果、计算特征和科学结论。创建于1986年,由瑞士生物信息学研究所和欧洲生物信息学研究所协同维护,截至2022年5月25日的2022_02 版中共收录567 483 个序列条目,包含204 940 973 个氨基酸,数据量逐年增加。此数据库中所提供的蛋白质序列信息十分详细,涵盖了蛋白质的功能、二级和四级结构、蛋白质翻译后修饰、蛋白质缺陷等信息,避免了序列的冗余,与其他数据库的交叉引用较为便利,方便用户检索。SWISS-PROT 数据库的增长如图5所示。

图5 SWISS-PROT 数据库的增长情况(图片来源于https://web.expasy.org/docs/relnoted/relstat.html)
布鲁克海文蛋白质数据库(PDB,https://www.rcsb.org/)创建于1973年,由美国国家科学基金等组织提供资助,是经实验测定的生物大分子三维结构数据库,利用Mol* 3D Viewer 可进行结构查看,如图6所示,其内容主要包括生物大分子的原子坐标、参考文献、一级和二级结构信息等。截至2022年,PDB 中可使用的蛋白质条目总数为190 404,每年发布的结构数量为4 917 条。

图6 4Z35 的3-D 结构信息(图片来源于PDB 数据库)
SCOP(https://scop.mrc-lmb.cam.ac.uk/)数据库由人工检查创建并由一系列自动化方法支持,旨在提供所有结构已知蛋白质之间的结构和进化关系的详细描述。因此,它提供了对所有已知蛋白质折叠的广泛调查、任何特定蛋白质近亲的详细信息,以及未来研究和分类的框架。截至2022-04-29的最新更新包括代表849 788 个蛋白质结构的72 082 个非冗余结构域。
CATH(https://www.cathdb.info/)数据库是一个免费、公开的在线资源,提供有关蛋白质结构域进化关系的信息,是蛋白质结构分类数据库。CATH 数据库版本众多,最新的CATH-Plus(v4.3)在CATH 中可用的核心分类信息基础上添加了大量数据。CATH-Plus 发布过程除了添加大量结合蛋白质结构、序列和功能的信息外,还包括许多手动注释检查,包括500 238 个域、5 481 个超家族、150 885 个带注释的PDB、82 665 284 个蛋白质序列。因此,在CATH-Plus 中可以获得更深入的信息。
3 结论
目前,蛋白质结构的预测方法呈现出多样化,尤其是在计算机理论飞速发展的背景下,蛋白质结构的预测理论和技术取得了很大的进步,但在预测精度和序列信息的提取等方面仍存在不足。
若要提高蛋白质结构的预测性能,需要在以下几个方面进行深入的研究:(1)降低蛋白质序列中信息的冗余度;(2)随着蛋白质氨基酸序列长度的急剧增加,蛋白质结构预测的精度有所下降,提高预测方法对数据处理速度和预测精度的重要性日益凸显;(3)目前的蛋白质数据库能否提供足够的有关结构的信息;(4)面对蛋白质结构与序列数据间的巨大差距,如何设计更高效的算法;(5)有些算法仍停留在理论研究层面,预测精度还无法达到实际应用的水平,更无法解答蛋白质结构中的折叠现象;(6)对分类算法的挖掘还不够深入,需要寻找更普遍适用的机器学习算法。
蛋白质结构预测在蛋白质功能研究中具有举足轻重的地位,现代科学技术的飞速发展使得蛋白质结构的预测仍然是一个值得深入探讨的重要课题。
