TET酶家族在神经胶质瘤干细胞中的分化调控机制及其潜在治疗靶点的研究进展
2022-10-20傅雨昂魏子龙
傅雨昂 魏子龙
(1上海市浦东医院-复旦大学附属浦东医院神经外科 上海 201399;2复旦大学公共卫生学院2018级预防医学本科生 上海 200032)
TET酶 家 族 蛋 白 由TET1、TET2和TET3组成,都含有一个保守的双链β-螺旋结构域、一个富含半胱氨酸的结构域以及辅助因子Fe(Ⅱ)和α-酮戊二酸的结合位点,共同形成C端核心催化区域,TET1和TET3还有一个n端CXXC锌指结构域,可以与DNA结合[1]。TET酶家族具有将5-甲基胞嘧啶(5-methylcytosine,5mC)转化为5-羟甲基胞嘧啶(5-hydroxymethylcytosine,5hmC)的能力,进而调控DNA甲基化[2],不仅调控细胞分化[3],而且在多种病理生理过程中发挥重要作用[4]。自TET酶家族蛋白质被发现以来,在神经胶质瘤以及其他恶性肿瘤中也发现了5 mC氧化异常和TET酶家族蛋白功能失调[5-7]。近年来,关于5mC氧化异常和TET酶家族蛋白功能失调对神经胶质瘤的影响得到广泛的研究[8-10]。
本文对TET酶家族在神经胶质瘤干细胞(glioma stem cell,GSC)分化过程中,通过去甲基化调控基因表达的具体机制及其在DNA损伤应答和修复过程的作用加以阐述,并对干预TET酶家族及其上游异柠檬酸脱氢酶(isocitrate dehydrogenase,IDH)基因的功能治疗神经胶质瘤面临的挑战进行分 析 讨 论 。 突 变IDH的 拮 抗 剂(mut-IDHantagonist)有望成为新的胶质瘤治疗策略,介导缺氧条件下DNA修复相关的TET1和TET3成为研究热点,或将成为新的治疗突破点。
GSC的研究现状肿瘤干细胞(cancer stem cell,CSC)是具有正常干细胞自我更新、多向分化潜能和无限增殖能力特性的肿瘤细胞[11]。依据CSC的分子特征,靶向性凋亡CSC或诱导CSC有效分化,是有效控制肿瘤复发、转移和耐药的手段之一。
GSC是神经系统肿瘤组织中具有干细胞基本特性的一部分细胞,包括具备自我更新能力、无限增殖能力和多向分化潜能的细胞[12-13],是CSC的热点研究模型之一。GSC的起源假说之一是正常神经干细胞(neural stem cell,NSC)在聚积基因突变、DNA损伤和基因组重排导致的染色体不稳定和表观遗传修饰混乱后可转化成为CSC[14-15]。GSC起源至今无明确定论,当前研究主要围绕NSC肿瘤性转化学说、成熟胶质细胞失分化学说、融合细胞学说和水平基因传递学说等四类学说展开:NSC肿瘤性转化学说认为,GSC可能来源于与之生物学特性相似、基因标志物相同的NSC,或在特定的环境刺激下通过基因改变获得自我更新能力的干祖细胞(stem and progenitor cell)[16-19];成熟胶质细胞失分化学说则认为其由胶质细胞通过失分化、基因突变、基因重排及遗传改变等方式再次激活,从而具有更强 的 增 殖 能 力 转 变 而 来[16,20];融 合 细 胞 学 说 认 为GSC的来源可能是促融合因子介导的融合细胞[21];水平基因传递学说则认为其由胶质细胞通过细胞吞噬、吞饮实现与其非子代细胞之间遗传物质的传递,使某些致病基因可在不同的细胞之间传递而产生[22]。GSC起源于某种细胞还是几种细胞的合体,NSC转化为GSC的调控机制和转化发生的区域在脑室下区域还是脑室外区域尚不明确[23]。
GSC的不对称分化导致其具有高度异质性的特点。早 在2010年,Verhaak等[25]便获得了肿瘤间异质性的证据,根据不同患者肿瘤样本中的基因突变和表达谱将胶质母细胞瘤(glioblastoma multiforme,GBM)分为4种亚型,其中2种亚型有助于GBM治疗后的异常生长和肿瘤复发[24-25]。2014年,Sottoriva等[26]通过单细胞测序发现,同一肿瘤组织的不同区域呈现不同的分型,称为肿瘤内异质性,并且不同亚群的GSC对不同的表观修饰抑制剂具有特异的敏感性[27]。这些研究揭示了不仅肿瘤组织异质性会影响治疗手段和效果,肿瘤组织GSC所具有的表观修饰异质性也将成为基础研究和临床精准治疗的关键因素。
TET酶家族的研究现状TET酶家族蛋白质具有保守的富胱氨酸结构域和双链β螺旋结构域,这是依赖Fe(Ⅱ)和α-酮戊二酸的双加氧酶的催化中心。TET酶家族蛋白质具有独特的酶促功能,可促进DNA去甲基化过程。TET介导5mC氧化,是将其转化为5hmC的双加氧酶,并进一步氧化产生5-甲酰基胞嘧啶(5-formylcytosine,5fC)和5-羧基胞嘧啶(5-carboxylcytosine,5caC)[28-31],胸腺嘧啶DNA糖苷酶能够氧化5fC和5caC,再经过碱基切除修复得到未被修饰的胞嘧啶。
TET酶介导的DNA去甲基化受到多种辅助因子和代谢物的调节。TET酶的作用依赖于α-KG和Fe2+。α-KG是三羧酸循环的重要中间体,推测能够通过降低α-KG的代谢活动抑制TET酶介导的去甲基化作用,当IDH1和IDH2发生突变时,α-KG无法正常合成,已有的α-KG被转化为2-羟基戊二酸(2-hydroxyglutarate,2HG),2HG是α-KG的类似物,能与α-KG竞争抑制TET蛋白。Fe2+则能增强TET的 催 化 活 性[32]。 锌 指 转 录 因 子(zinc finger transcription factors,ZSCAN)可募集TET酶家族,从而调控包括重编码在内的多种细胞过程,锌指转录因子成员中的Zscan4f是TET2调控靶基因和促进诱导多能干细胞(induced pluripotent stem cell,iPSC)生成的重要伙伴[33]。TET酶家族中TET2易位突变最早在急性髓系白血病中被发现,这一突变导致TET2丧失去甲基化功能。TET酶家族的抑癌机制除了通过去甲基化途径调控基因表达,还可能参与了DNA损伤修复过程。
TET酶家族的3个成员参与DNA损伤应答和修复过程的现象及机制研究分别被报道过。TET1通过ATM依赖的方式对DNA损伤产生响应,从而在浦肯野细胞内产生大量的5hmC修饰,缺失TET1会影响损伤修复的激活,导致细胞凋亡[34]。TET2有助于5hmC的富集,一种通过与共激活因子和序列特异性DNA结合因子的三元相互作用将TET2靶向到特定启动子:Smad核内相关蛋白1(Smad nuclear interacting protein 1,SNIP1)沟 通TET2与包括c-myC在内的多种转录因子的相互作用;TET2以SNIP1依赖的方式保护细胞免受DNA损伤诱导的凋亡;TET2-SNIP1-c-myC轴支持TET2的DNA序列特异性募集。TET2-SNIP1-cmyC通路介导DNA损伤修复反应,保护细胞免受DNA损伤诱导的凋亡,从而将表观遗传控制与基因组稳定性的维持联系起来[35]。TET3产生5hmC参与损伤响应则被认为是受到ATR磷酸化的调控,并促进DNA去甲基化和5hmC的积累[36],从而起到DNA修复与基因组稳定性的维护作用[37]。与TET2类似,TET3的催化活性对DNA正常修复和细胞存活也很重要。有研究曾在TET2和TET3双敲除的白血病模型中观察到非同源末端修复(nonhomologous end joining,NHEJ)和 同 源 重 组 修 复(homologous recombination,HR)的基因普遍受到转录抑制。
有研究认为TET酶在细胞复制过程中有着维持基因组完整性的作用。活性DNA去甲基化可能特异性发生在DNA损伤部位,这一事实表明TET酶和/或5hmC可能在DNA修复中起 直 接 作用[38]。5hmC参与的表观遗传调控的缺失可能有助于疾病进展[39-40]。5hmC可能具有DNA损伤的表观遗传标记的作用,并被证明可以促进基因组的稳定性。
5hmC在DNA修复中的潜在作用亟待研究,5hmC标记可出现在DNA损伤部位的DNA共价修饰,研究TET活化与5hmC的产生是否与DNA损伤类型有关至关重要。5hmC可能是TET在DNA损伤部位激活的副产物,这是清除5mC所必需的。还有一种可能,即对主导DNA修复的是TET酶,而不是5hmC。确认DNA损伤介导的5hmc识别因子与TET酶家族相互作用的关系,对于理解5hmC和TET蛋白在DNA修复中的潜在作用至关重要[36]。
胶质瘤及GSC中涉及到的TET酶家族的研究情况TET酶家族的抑癌机制除了通过去甲基化途径调控基因表达,可能还参与了DNA损伤修复过程。在TET酶家族中存在潜在的劳动分工。
已有研究报道,神经胶质瘤中的整体低甲基化和位点特异性异常甲基化以及其他表观遗传修饰是神经胶质瘤进展过程中基因组不稳定的重要因素。在以缺氧介导的神经胶质瘤细胞DNA去甲基化过程中,发挥作用的去甲基化酶是TET1和TET3,而不是TET2。TET1和TET3与八聚体结合转录因子4(octamer-binding transcription factor 4,OCT4)和转录因子NANOG有活性的调控区域直接结合去甲基化酶TET1和TET3。从GSC中敲除去甲基化酶TET1和TET3时,干细胞基因OCT4和NANOG的表达均下降,并抑制神经球的形成,在缺氧状态下TET1和TET3的去甲基化是OCT4和NANOG过表达的机制,有助于胶质瘤中GSC的形成[41]。TET1和TET3在低氧条件下各自独立调节多能性相关基因和分化相关基因。
TET2在 产 生5fC和5caC方 面 作用 更 强[30],尤其是增强子上的5hmC富集所必需的[42]。TET2的表达还与GSC对DNA损伤后的修复相关。GSC比NSC表达更高水平的DNA修复基因,这可能是其对辐射或化疗诱导的DNA损伤抗性的原因之一。使用siRNA敲除TET2可使GSC细胞系对博莱霉素治疗敏感,并且TET2表达的GSC越高则越能抵抗博莱霉素造成的DNA损伤并继续增殖,表明TET2确实能促进DNA损伤后的修复。TET2也可能在细胞周期中催化生成和DNA损伤应答相关的基因启动子上的5fC/5caC,这些基因在GSC中表达高于NSC,且与TET2水平呈正相关,提示TET2可能调控这些位点的表达。
TET3蛋白同时分布于细胞核和细胞质内,与GSC中 分 化 表 型(glial fibrillary acidic protein,GFAP)基因的表达高度相关。人们通过研究GSC核受体TLX(NR2E1)和TET3的级联调控机制发现TET3可以抑制GSC的生长[43]。TLX是一种可以抑制TET3表达的转录因子,TET3在TLXTET3轴中处于TLX的下游,可以抑制GSC的生长、自我更新和肿瘤发生。然而,TET3一方面受到TLX调控,抑制GSC的生长,另一方面却可以通过其他通路增加GSC的致瘤性。GCS的细胞外基质(extracellular matrix,ECM)通过层黏素-整合素 α6上调TET3蛋白。TET3反过来介导DNA胞嘧啶5'-羟甲基化(5hmC)的表达,并上调对维持GSC必要生命活动至关重要的基因。激活整合素α6-局部黏着斑激酶通路(integrin α6-focal adhesion kinase pathway)可提高GSC中信号传导与转录激活因子家 族(signal transduction and activators of transcription,STAT)的 成 员STAT3活 性 ,增 强TET3表达,从而提高5hmC水平。此外,STAT3直接调控TET3的表达,这两个蛋白质在GSC集群中 与5hmC共 定 位 。 因 此 ,ECM-整 合 素 α6-STAT3-TET3轴调控了GSC重要基因的羟甲基化,从而增加了GSC的致瘤性[44]。
此外,GSC中TET高表达可能会增加化疗耐药性。GSC在肿瘤中以小亚群形式存在,但积极地促进了整体放疗和化疗耐药以及肿瘤复发。在多个人源性组织异种移植(patient-derived xenograft,PDX)GSC中分析了全基因组DNA表观遗传修饰、增强子标记和转录组,发现在GSC中TET表达缺失与5mC和5hmC的整体损失和5fC/5caC的增加相关,与NSC相比,TET表达在GSC分化过程中有不同的反应,TET3亚细胞定位与分化能力有关;分化诱导的5mC和5hmC重编码定位于不同的增强子区域,有助于发育基因的调控。整合素α6-STAT3-TET3轴本身也可以增加GSC的耐药性。GSC中TET2高水平表达有助于化疗耐药,可能是通过调节DNA损伤反应和修复系统来实现。
干预TET酶家族治疗神经胶质瘤的展望TET1活性缺失与多种肿瘤的进展、转移和患者生存率低直接相关,因此TET酶家族通常被认为是抑制肿瘤发生发展的抑癌基因的重要表达产物之一。
IDH突变干扰TET酶家族的功能,其所造成的胶质瘤CpG岛甲基化表型(cytosine-phosphateguanine island methylator phenotype,G-CIMP)与GBM亚型有关,包括在全基因组范围内调节甲基化模式,改变转录程序和改变分化状态[45]。当TET酶功能发生紊乱时,产生的5hmC水平降低,同时缩短患者生存期[46]。因此,TET介导的DNA去甲基化过程及其表观修饰产物是治疗和精准诊断GBM的关键切入点(图1)。
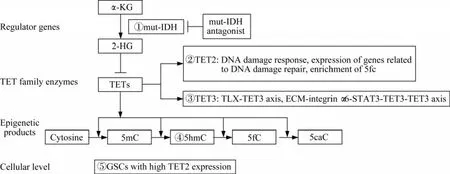
图1 干预TET酶家族治疗神经胶质瘤的治疗靶点示意图(治疗靶点①~⑤)Fig 1 Schematic diagram of therapeutic targets for intervention of TET family enzymes in the treatment of glioma(therapeutic target①-⑤)
TET2在GSC中的表达高于NSC,TET2的表达与修复基因的表达呈线性相关。且TET2与DNA损伤响应、修复相关基因的表达及其启动子甲酰化胞嘧啶(5fC)的富集显著相关,从而成为一种潜在的治疗靶点。明确GSC中独特的表观基因组特征和相关的基因网络,针对这些特征可以在不损伤邻近正常细胞的情况下应用于临床靶向治疗[47]。
调节TET3相关通路可以为治疗GBM开辟新的思路。调节ECM-整合素α6-STAT3-TET3轴可以逆转通常与CSC增加有关的耐药性[44]。调节TLX-TET3轴,抑制核受体TLX的表达可抑制GBM的 生 长[43]。 病 毒 载 体 递 送 的TLX shRNA或纳米载体递送的TLX siRNA治疗人类GSC移植小鼠,可抑制肿瘤发展并延长生存期。
GSC在转录、DNA修饰和增强子标记等多个水平上存在异质性,这些异质性也部分反映在GSC间TET表达水平上。高TET2表达的GSC可能是临床上抗药物治疗的靶点。GSC特异性特征可能赋予该细胞群独特的分子谱和表观遗传特性,并作为抑制或消除GSC群体的潜在治疗靶点[41]。
有研究表明,在体外用谷氨酸替代IDH1 132残基上的精氨酸会造成TET酶活性下降[48]。突变未必导致IDH1蛋白失活,例如阻止使用异柠檬酸作为底物的突变蛋白质,可以允许酶使用其他未知底物,从而使得酶并未完全失活。如果未来的研究证实了这种可能性,突变的IDH基因可能成为治疗干预的目标。
结语综上所述,TET酶家族与神经胶质瘤发生相关,TET介导的DNA去甲基化过程及其表观修饰产物是治疗和精准诊断GBM的关键切入点。确定神经GSC中独特的表观基因组特征和相关的基因网络以及TET2对基因网络的调控机制,可以在不损伤邻近正常细胞的情况下应用于临床靶向治疗。虽然已对TET2基因突变进行了一系列深入研究,但仍需要考虑TET2能否像其他基因一样作为神经胶质瘤的危险分层因子。调节TET3相关通路可以降低GSC的致瘤性,抑制肿瘤生长。GSC在转录、DNA修饰和增强子标记等多个水平上的特异性特征可能赋予该细胞群独特的分子谱和表观遗传特性,作为抑制或消除GSC群体的潜在治疗靶点。IDH的突变是脑胶质瘤CIMP的分子基础,其所造成的基因组高甲基化状态有助于改变转录程序和分化状态,为理解神经胶质瘤的发生提供了框架,可能成为治疗干预的目标[49]。近年来随着IDH1/2突变的致癌功能及其相关代谢产物顺-2-羟基戊二酸、缺氧反应和DNA修复研究逐步深入,突变IDH的拮抗剂有望成为新的胶质瘤治疗策略,与介导缺氧反应下DNA修复相关的TET1和TET3可能成为新的突破点。
作者贡献声明傅雨昂 文献调研和整理,论文撰写和修订。魏子龙 论文指导和修订。
利益冲突声明所有作者均声明不存在利益冲突。
