Two-dimensional g-C3N4 Compositing with Ag-TiO2 as Deactivation Resistant Photocatalyst for Degradation of Gaseous Acetaldehyde
2022-09-29XUEHongyunWANGCongyuMAHMOODAsadYUJiajunWANGYanXIEXiaofengSUNJing
XUE Hongyun, WANG Congyu, MAHMOOD Asad, YU Jiajun, WANG Yan, XIE Xiaofeng, SUN Jing
Two-dimensional g-C3N4Compositing with Ag-TiO2as Deactivation Resistant Photocatalyst for Degradation of Gaseous Acetaldehyde
XUE Hongyun1,2, WANG Congyu1, MAHMOOD Asad1, YU Jiajun1,2, WANG Yan1, XIE Xiaofeng1, SUN Jing1
(1. State Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China; 2. University of Chinese Academy of Sciences, Beijing 100049, China)
The photocatalysts deactivation is one of the major issues, which lowers the usefulness of photocatalytic oxidation technology for the removal of low content volatile organic compounds (VOCs). Here, we carried out a series of experiments to demonstrate that the photocatalysts stability could be significantly improvedcoupling the oxide base semiconductors,, TiO2with 2D materials such as graphitic carbon nitride (g-C3N4). Initially, when Ag modified TiO2(AT) was used for the gaseous acetaldehyde degradation, a robust deactivation was observed within 60 min. The AT catalyst completely lost its activity when the reaction time was extended to 400 min. On the contrary, the g-C3N4modified AT (CAT) showed superior photocatalytic performance and improved stability (600 min). TheFT-IR, PL, and photocurrent studies suggested that the accumulation of reaction intermediates in the case of AT fundamentally caused the deactivation. However, the g-C3N4provided excessive adsorption sites for the reaction by-products which improved the stability. Additionally, the PL and ESR studies suggested that the existence of g-C3N4improved the charge separation and production of reactive oxygen species, which facilitated the photodegradation of acetaldehyde and ultimate reaction products. This study realizes the usefulness of 2D materials for developing stable and visible light active photocatalysts for applications in sustainable VOC abatement technology.
photocatalysis; titanium dioxide; g-C3N4; deactivation-resistant;diffuse reflectance infrared Fourier transform spectroscopy
Nowadays, people spend much of their time (>80%) indoors, which means that indoor air quality needs special attention[1]. The low boiling point volatile organic compounds (VOCs) are one of the major indoor air pollutants,, aromatic hydrocarbons and aldehydes. It has been well thought out that VOCs may cause some serious health problems and environmental concern[2-3]. The photocatalytic oxidation technology (PCO) is regarded as a promising method to eliminate the low content VOCs in the indoor air[4]. TiO2is widely used in heterogeneous catalysis due to its excellent chemical stability, strong oxidizing power, and low-cost[5]. Researchers have done a lot of work regarding TiO2modification to achieve better catalytic activity[6-10]. However, the deactivation of TiO2in the gas-solid photocatalytic reaction is a serious problem, which has been given relatively less attention that seriously undermines the practicality of PCO technology in the VOCs abatement regime.
The deactivation of catalysts maybe due to the structural changes or the surface coverage by intermediates and coke deposition[11-12]. The deactivation of photocatalysts is generally the second type. Previously, it has been reported that toxic by-products produced in the photocatalytic oxidation process would be deposited on the surface of TiO2, leading to the deactivation[13]. The refractory intermediates not only inhibit the light absorption of photocatalysts, but also obstruct the binding of reactants and active surface sites. Some post-processing methods have been developed to solve the deactivation problems, such as UV lamp irradiation, heat treatment, aqueous solution washing,.[14-16]. But these methods are neither convenient nor economical. The best strategy should be to prevent the photocatalysts deactivation. Recently, the deactivation-resistant photocatalysts can be obtained by the following methods: i) Morphology Control. Weon.[17]synthesized TiO2nanotubes (TNT) structure, which allowed oxygen molecules to bind to the active sites easily and facilitated the decomposition of organic pollutants. ii) Surface Modification. Weon.[18]promoted the formation of removable hydroxyl radicals by surface fluorination, solving the problem of deactivation of Pt-TiO2catalysts. iii) Surface Oxygen Vacancies. Dong.[19]constructed the BiOCl with oxygen vacancies which could generate more free radicals with high oxidative capacity, shorten the degradation pathway of toluene and effectively inhibit the formation of toxic intermediates.
Two-dimensional materials have a remarkable potential as pollutant molecular sensing materials due to their layered structure and high adsorption property[20]. When composited with photocatalysts, two-dimensional materials can effectively promote the separation of photogenerated carriers and the adsorption of target pollutants[21]. Our group recently founded that the rGO/Er3+-TiO2composite could significantly improve the VOCs adsorption and resist deactivation[22]. Two-dimensional materials can not only effectively adsorb target pollutants, but also provide extra adsorption sites for the intermediates. Additionally, it can facilitate the light harvesting and charge separation properties of the composite framework.
Recently, we found that the Ag decoration can improve the photocatalytic activity of TiO2. However, the degradation efficiency of the Ag-TiO2sample gradually decreased with the passage of time. When the Ag-TiO2system was compounded with two-dimensional g-C3N4, good photocatalysts stability was achieved[23]. In this work, we verified the reason of deactivation of the Ag- TiO2sample, which was due to the successive accumulation of intermediates. The possible photocatalytic degradation pathway of acetaldehyde was inferred by theDRIFTS. The effect of intermediates and the role of g-C3N4in photocatalysts anti-deactivated were proposed by a series of tests.
1 Experimental
1.1 Chemicals
All chemical agents used in experiment were analytical reagents and without further purification. TiO2(Degussa P25) was bought from Evonik Industries AG, Germany. Melamine (AR, 99.0%), NaOH (AR, 96.0%) and AgNO3(AR, 99.8%) were purchased from Sinopharm Chemical Reagent CO., Ltd, China. NaBH4(AR, 98%) and 5,5-dimethyl-1-pyrroline--oxide (DMPO, AR, 97%)were purchased from Aladdin Reagent Co., Ltd, China.
1.2 Preparation of samples
1.2.1 Synthesis of g-C3N4
The graphite carbon nitride (g-C3N4) was synthesized by using a high temperature thermal polymerization reaction[24]. For instance, melamine was calcined at 550 ℃ for 4 h with a heating rate of 10 ℃/min in a Muffle furnace. Yellow powder was achieved after naturally cooling down to ambient temperature, hereafter, designnated as CN.
1.2.2 Synthesis of Ag-TiO2
The Ag-TiO2composites were prepareda chemical reduction method by adjusting pH[25]. Initially, TiO2(1.2 g) was dispersed in 100 mL deionized water. Subsequently, 0.1 mol/L NaOH solution was added to the suspension under constant stirring to adjust pH to 8.5. Next, 0.56 mL of 0.2 mol/L AgNO3solution was added to the prepared suspension and kept stirring for 1 h in the dark to achieve the adsorption equilibrium of Ag+. Ar was injected into the suspension and stirred at ambient temperature for 30 min. Finally, 5.6 mL of 0.1 mol/L NaBH4/NaOH mixture solution was slowly added under constant stirring for another 30 min. Finally, the sample was centrifuged and dried to obtain gray powder, designated as AT.
1.2.3 Synthesis of g-C3N4/Ag-TiO2
The g-C3N4/Ag-TiO2composites were prepared by electrostatic adsorption. Typically, g-C3N4and Ag-TiO2powders (mass ratios of 1 : 1) were dispersed in deionized water for 10 h, using an ultrasonic bath. The as-prepared mixture was calcined at 400 ℃ for 1 h (heating rate of 2 ℃/min) to obtain yellow powder, designated as CAT.
1.3 Characterization
The X-ray diffraction (XRD, D8ADVANCE, Bruker, Germany) used Cu Kα radiation (=0.154 nm, 2varied from 2° to 80°, step of 5 (°)/min) to characterize the crystalline structure of the materials. The photoluminescence (PL) spectra were acquired on the LS-55 fluorescence spectrophotometer (PerkinElmer, USA) with an excitation wavelength of 320 nm. The photocurrent was tested by using the CHI660D Electrochemical Workstation (China) with a three-electrode quartz cell. Electron spin resonance (ESR) signals were measured by the JES-FA200 electron paramagnetic resonance spectrometer (JEOL, Japan). DMPO was used as radical trapper.
1.4 Photocatalytic experiments and intermediates detection
Acetaldehyde gas was selected as a model VOC to evaluate the photocatalytic activity and durability. The tests were carried out with a continuous flow-phase gas in an automated measurement setup, which contained a gas mixer, gas flow monitoring system and a gas chromatograph. The 400 W Xenon lamp with a 420 nm cut-off filter was used as the light source to estimate the performance of photocatalysts under the visible light irradiation. The concentration of acetaldehyde was 25×10–6obtained by mixing acetaldehyde gas (50×10–6) with air in the ratio of 1 : 1. The flow rate was set to 20 standard cubic centimeter per minute (sccm). The degradation ratio () of acetaldehyde was calculated by= (1 –/0)×100%, in which0is the initial concentration, whileis the real-time concentration of acetaldehyde.
The intermediates of the photocatalytic reaction were analyzed bydiffuse reflectance infrared Fourier transform spectroscopy (DRIFTS, IRTracer-100, Shimadzu Co., Ltd, China). 30 mg sample was put into a mold containing a nickel mesh and inlet high pure nitrogen. After 20 min, the reactor was heated to 300 ℃ and maintained for 30 min to clean the gas pipes and the catalyst surface. Next, gaseous acetaldehyde (25×10–6) was allowed to flow at room temperature in the reaction cell. After subtracting the background spectrum, the adsorption spectra were collected accordingly using a spectral acquisition software. The adsorption was carried out for 120 min in the dark. Next, the samples were irradiated using the Xenon lamp (CHF-XM-250W) to degrade the target VOC. The DRIFTS data was obtained every 3 min to trace the organic functional groups/ intermediates formation on the catalyst surface.
2 Results and discussion
Fig. 1(a, b) show the photocatalytic degradation curves of acetaldehyde under visible light irradiation for different catalysts. Compared with AT, the adsorption properties and photocatalytic degradation efficiency of the ternary composites CAT were greatly improved. The photocatalytic degradation efficiency of AT and CAT was 41.2% and 69.5%, respectively. Moreover, the AT sample showed a rapid deactivation. For instance, the degradation efficiency began to decrease after 60 min. The mineralization curves further complement the degradation results (inset in Fig. 1(a)). The CO2production was observed to decrease after 60 min and the catalyst surface turned yellow with reaction time increasing. In around 400 min, the degradation efficiency of the AT dropped to 0. However, the CAT composites maintained a stable degradation performance up to 600 min, under the visible light irradiation.
To trace the possible reason of catalyst deactivation, we first performed the structural characterization. From the XRD patterns (Fig. 1(c)), the characteristic peaks located at 2=25.4° (101), 37.8 ° (004), 48.1° (200), 53.9° (105), 55.1° (211), 62.7° (204), 68.7° (116), 70.3° (220) and 75.1° (215) confirmed the anatase TiO2, while the peaks located at 2=27.4° (001), 36.1° (101), 41.3° (111) and 54.4° (211) were assigned to rutile TiO2. g-C3N4showed two characteristic peaks located at 2=13.1° and 27.5°, corresponding to the (100) lattice facet of the g-C3N4triazine structural unit and (002) lattice facet of the graphite phase aromatic segment, respectively. The structure of both samples did not change after the photodegradation reaction. It can be inferred that the deactivation of the AT sample could not be associated with the structural changes.
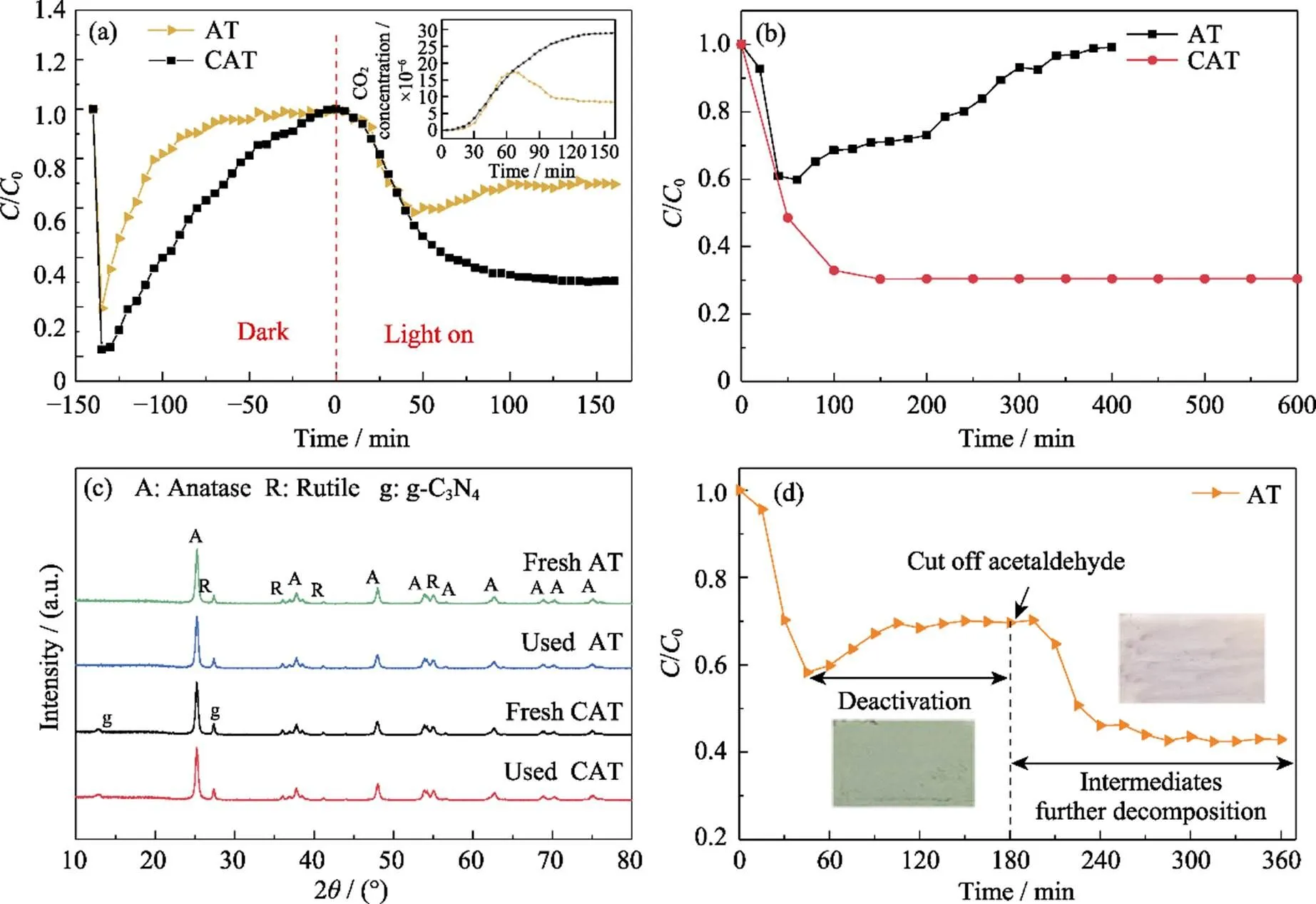
Fig. 1 Activation maintained by CAT for gaseous acetaldehyde
(a) Adsorption, photodegradation and CO2generation curves of acetaldehyde gas by AT and CAT samples; (b) Photocatalytic degradation of acetaldehyde by AT sample under visible light irradiation for 400 min and CAT sample under visible light irradiation for 600 min; (c) XRD patterns of AT and CAT samples before and after degradation of acetaldehyde gas (160 min) under visible light irradiation; (d) Photocatalytic degradation of acetaldehyde by AT sample under visible light irradiation for 360 min, and cutting off acetaldehyde inlet at 180 min
To comprehend the deactivation of AT composites, a new AT sample was used to degrade acetaldehyde under the visible light irradiation. As shown in Fig. 1(d), the catalyst began to deactivate after 60 min and the surface turned yellow. The degradation efficiency reduced to 30% after 180 min. The yellow species on the surface were more likely the intermediates of acetaldehyde oxidation. At this stage, the flow of acetaldehyde was stopped while keeping the light on for 3 h. In this way, the catalyst recovered activity and the yellow color disappeared, which suggested that the accumulated intermediates were further degraded. The accumulation of acetaldehyde oxidation products would cover the active surface sites, which might be the reason for the AT sample deactivation. However, the CAT sample can prevent deactivation and maintain a stable and efficient degradation of acetaldehyde. This suggested that the g-C3N4play an important role in maintaining the activity of the photocatalyst.
TheDRIFTSspectra were further used to analyze the acetaldehyde adsorption and degradation process. As shown in Fig. 2 and Tables S1 and S2, the peaks at 1732, 1716, 1699 cm–1were assigned to(C=O), which was attributed to acetaldehyde adsorption on TiO2surface. The bands located at ~1620 cm–1were attributed to the(C=C) stretching vibrations of crotonaldehyde, which was formed by aldol condensation reaction of acetaldehyde adsorbed on TiO2[26]. The bands at 1699 and ~1315 cm–1were assigned to acetic acid, while the bands at 1716, 1377 cm–1were assigned to formic acid. The bands at around 1541, 1558, 1458, 1473, 1419, 1339 cm–1were due to acetate species, while the bands at around 1541, 1558, 1377, 1315 cm–1were assigned to formate species. The peak at 1458 cm–1was identified as(CH2) of dioxymwthylene (DOM) species, which was formed by formaldehyde adsorbed on TiO2[27]. From the above results, the acetaldehyde degradation pathway could be inferred. In the adsorption process, the acetaldehyde adsorbed on TiO2surface was converted to 3-hydroxyl bualdehyde and coutaraldehyde groups by aldehyde condensation and dehydrogenation[26]. Small amounts of acetate and formate were also produced by the oxidation of acetaldehyde at the surface oxygen sites[28-29]. Further on, the acetaldehyde and the initially generated species were oxidized to acetic acid, formaldehyde and formic acid by active species (, •O2–and •OH) which was produced on the photocatalyst surface under the visible light irradiation. The formic acid and acetic acid adsorbed on TiO2reacted with metal atom Ti to form formate and acetate species, which were in dynamic equilibrium with acetic acid and formic acid[27]. The formaldehyde and DOM were also in steady-state equilibrium. Finally, theformate and acetate species were photocatalytically oxidized to carbon dioxide and water[30].
In order to further explore the influence of intermediates and the role of g-C3N4on the anti-deactivation, PL, photocurrent and ESR were carried out. The photocurrent spectroscopy and PL spectra were used to investigate the generation, separation and recombination of photogenerated charges during the degradation of acetaldehyde (Fig. 3). As shown in Fig. 3(a), the photocurrent intensity of AT sample increased from 0 to 180 min under the visible light irradiation, which meant that the transfer of photogenerated carries was inhibited. This may be due to intermediates deposited on the surface and occupied active sites, which captured electrons at the electrode/ electrolyte interface. After cutting off the inlet acetaldehyde while keeping the light on from 180 min to 300 min, the photocurrent intensity gradually decreased. It can be seen from the previous conclusion that keeping light on could decompose the surface sediments again, which further confirmed that the intermediates hindered the charge transfer. The results of PL (Fig. 3(c)) also supported this interpretation. In the case of illumination time from 0 to 180 min, the recombination rate of photogenerated carriers gradually increased. After cutting off the gas, the separation ability of carriers recovered. However, the CAT sample showed different results (Fig. 3(b, d)). The photocurrent intensity did not decrease and the PL intensity did not increase, which suggested that the separation efficiency of photogenerated electron-hole pairs did not get negative effect during the long reaction. This may be due to the intermediates accumulated on the 2D g-C3N4while retaining the original active sites. Instead, the deposited intermediates promoted the transfer and separation of the photogenerated electrons and holes, which may be because the intermediates were further carbonized into conductive coke[31].

Fig. 2 In-situ DRIFTS spectra of (a) AT and (b) CAT photocatalysts degrading acetaldehyde gas under visible light irradiation, and (c) photocatalytic reaction routes of acetaldehyde
The generation of superoxide radicals (•O2–) and hydroxyl radicals (•OH) were measured by the ESR (Fig. 4), which influenced the oxidation of acetaldehyde and its intermediates. The intensity of DMPO-•O2–and DMPO-•OH signals in AT sample (Fig. 4(a, b)) gradually weakened over time during the first 180 min degradation of acetaldehyde, which may be due to less generation of photogenerated charges. The intensity of •O2–and •OH radicals gradually increased after stopping acetaldehyde but continuing the illumination. These results implied that the intermediates were further decomposed and the active sites were exposed again. However, the •O2–and •OH radicals’ intensity of CAT sample (Fig. 4(c, d)) did not decrease within 300 min as the photocatalytic reaction continuing, but increased. The introduction of 2D g-C3N4can provide additional adsorption sites for intermediates and keep the catalyst maintaining high activity. The formation of coke promoted charges transfer, which increased the intensity of •O2–and •OH radicals. ESR results fitted with the results of PL spectra and photocurrent response.
Based on our holistic detection, a proper mechanism was proposed to explain the reason for the anti- deactivation of the CAT. As illustrated in Fig. 5, the intermediates such as acetate and formate would be produced in the degradation process of acetaldehyde. On one hand, they were further decomposed by the superoxide radicals (•O2–). On the other hand, the introduction of the two-dimensional material g-C3N4with good adsorption performance could provide additional adsorption sites for the intermediates and retained the active sites of the photocatalyst. Therefore, the CAT was resistant to deactivation.
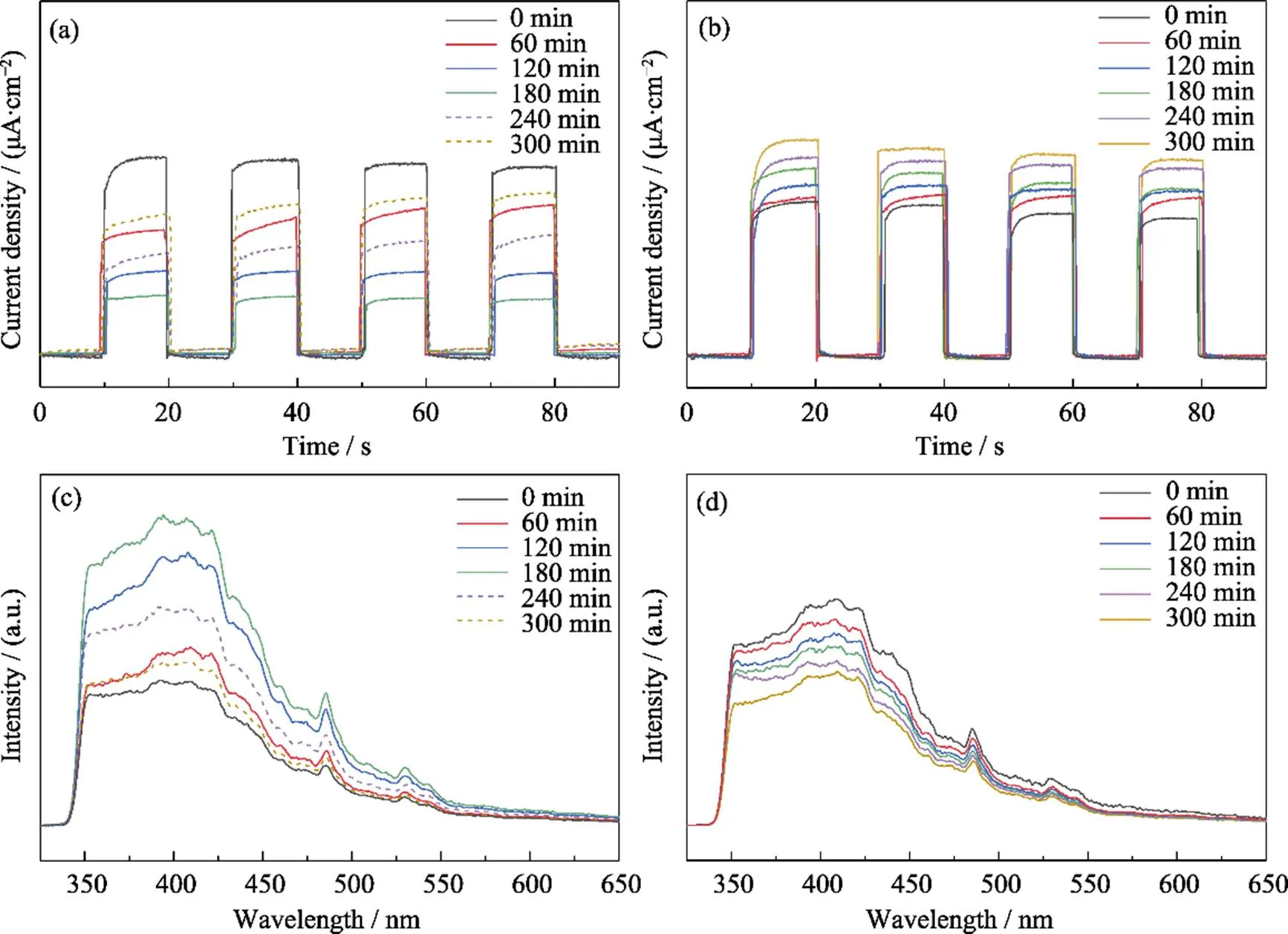
Fig. 3 Photocurrent and PL plots of the photocatalytic degradation of acetaldehyde
(a) Photocurrent and (c) PL plots of AT sample for the photocatalytic degradation ofacetaldehyde under visible light irradiation; (b) Photocurrent and (d)PL plots of the photocatalytic degradation of acetaldehyde by CAT sample undervisible light irradiation for 300 min 0, 60, 120, 180 min represent the photocatalytic reaction time of which the dotted lines of 240 and 300 min represent 1 and 2 h,respectively, when acetaldehyde was stopped but the light was kept on

Fig. 4 ESR profiles of (a, c) DMPO-•O2– and (b, d) DMPO-•OH for the photocatalyticdegradation of acetaldehyde by AT(a, b) and CAT (c, d) sample
Under visible light irradiation for 300 min0, 60, 120, 180 min represent the photocatalytic reaction time, of which the dotted lines of 240 and 300min represent 1 and 2 h, respectively, when acetaldehyde was stopped but the light waskept on
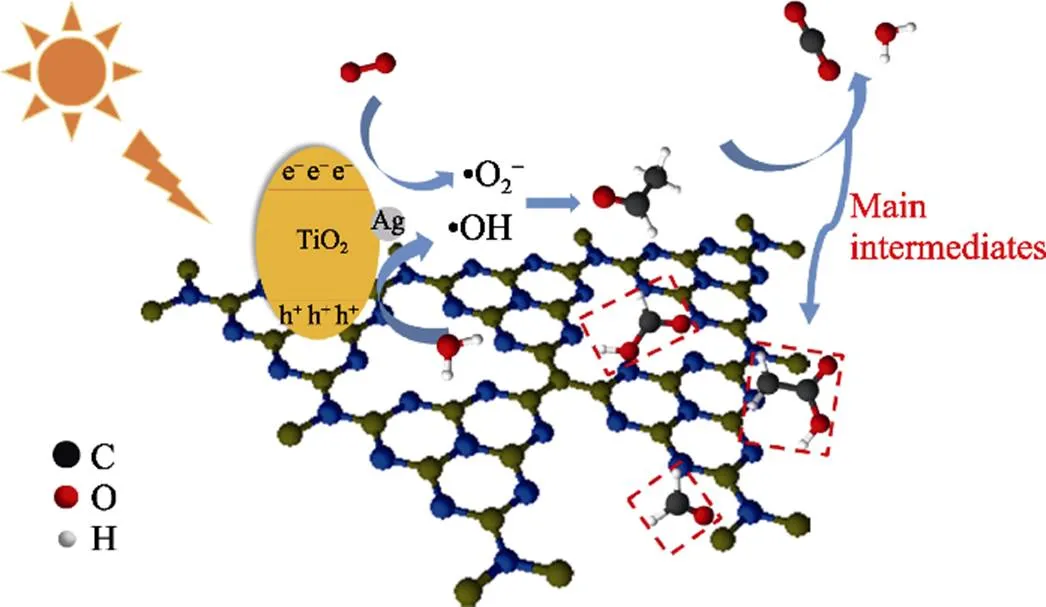
Fig. 5 Schematic of photocatalytic reaction mechanism
3 Conclusion
In summary, we explored the reaction path of photocatalytic degradation of acetaldehyde under the visible light irradiation and the mechanism of the deactivation of AT sample. In the degradation process of acetaldehyde, intermediates such as acetate and formate would be produced. The AT sample was easy to be deactivated due to the deposition of intermediates on the surface. The accumulation of intermediates on the photocatalyst surface gradually hindered the migration and dissociation of photo generated electron-hole pairs, which in turn inhibited the production of super-oxygen and hydroxyl radicals, ultimately leading to the deactivation of Ag-TiO2. However, the g-C3N4/Ag-TiO2had excellent adsorption property and photocatalytic degradation efficiency, and more could maintain high activity after reaction for 600 min, with excellent stability and durability. During the reaction, the active sites of CAT did not decreased. The introduction of the two-dimensional material g-C3N4with good adsorption performance could provide additional adsorption sites for the intermediates and retain the active sites of the photocatalyst, so that the CAT sample was resistant to deactivation. This work can provide a useful reference for the high-efficiency mineralization of VOCs and anti-deactivation methods in the photocatalytic process.
Supporting materials
Supporting materials related to this article can be found at https://doi.org/10.15541/jim20220123.
[1] WANG S B, ANG H M, TADE M O. Volatile organic compounds in indoor environment and photocatalytic oxidation: state of the art., 2007, 33(5): 694–705.
[2] ZHANG X Y, GAO B, CREAMER A E,Adsorption of VOCs onto engineered carbon materials: a review., 2017, 338: 102–123.
[3] HUANG R J, ZHANG Y L, BOZZETTI C,High secondary aerosol contribution to particulate pollution during haze events in China., 2014, 514: 218–222.
[4] KAMAL M S, RAZZAK S A, HOSSAIN M M. Catalytic oxidation of volatile organic compounds (VOCs)-a review., 2016, 140: 117–134.
[5] GUO Q, ZHOU C Y, MA Z B,Fundamentals of TiO2photocatalysis: concepts, mechanisms, and challenges., 2019, 31(50): 1901997.
[6] FU C, LI M J, LI H J,Fabrication of Au nanoparticle/TiO2hybrid films for photoelectrocatalytic degradation of methyl orange., 2017, 692: 727–733.
[7] DAI Y Q, COBLEY C M, ZENG J,Synthesis of anatase TiO2nanocrystals with exposed {001} facets., 2009, 9(6): 2455–2459.
[8] LI W, WANG F, LIU Y P,General strategy to synthesize uniform mesoporous TiO2/graphene/mesoporous TiO2sandwich- like nanosheets for highly reversible lithium storage., 2015, 15(3): 2186–2193.
[9] LIU H H, LI Y, XIANG M M,Single-layered MoS2directly grown on rutile TiO2(110) for enhanced interfacial charge transfer., 2019, 13(5): 6083–6089.
[10] LIANG H J, ZHANG B, GE H B,Porous TiO2/Pt/TiO2sandwich catalyst for highly selective semihydrogenation of alkyne to olefin., 2017, 7(10): 6567–6572.
[11] FORZATTI P, LIETTI L. Catalyst deactivation., 1999, 52(2/3): 165–181.
[12] ABBAS N, HUSSAIN M, RUSSO N,Studies on the activity and deactivation of novel optimized TiO2nanoparticles for the abatement of VOCs., 2011, 175: 330–340.
[13] MO J H, ZHANG Y P, XU Q J,Determination and risk assessment of by-products resulting from photocatalytic oxidation of toluene., 2009, 89(3/4): 570–576.
[14] HE C, CHENG J, ZHANG X,Recent advances in the catalytic oxidation of volatile organic compounds: a review based on pollutant sorts and sources., 2019, 119(7): 4471–4568.
[15] YANG X J, SUN H W, LI G Y,Fouling of TiO2induced by natural organic matters during photocatalytic water treatment: mechanisms and regeneration strategy., 2021, 294: 120252.
[16] HUANG H T, FENG J Y, ZHANG S,Molecular-level understanding of the deactivation pathways during methanol photo- reforming on Pt-decorated TiO2., 2020, 272: 118980.
[17] WEON S, CHOI W. TiO2nanotubes with open channels as deactivation-resistant photocatalyst for the degradation of volatile organic compounds., 2016, 50(5): 2556–2563.
[18] WEON S, KIM J, CHOI W. Dual-components modified TiO2with Pt and fluoride as deactivation-resistant photocatalyst for the degradation of volatile organic compound., 2018, 220: 1–8.
[19] DONG X A, CUI W, WANG H,Promoting ring-opening efficiency for suppressing toxic intermediates during photocatalytic toluene degradationsurface oxygen vacancies., 2019, 64(10): 669–678.
[20] ZONG B Y, XU Q K, LI Q J,Novel insights into the unique intrinsic sensing behaviors of 2D nanomaterials for volatile organic compounds: from graphene to MoS2and black phosphorous., 2021, 9: 14411–14421.
[21] REN Y, DONG Y Z, FENG Y Q,Compositing two- dimensional materials with TiO2for photocatalysis., 2018, 8(12): 590.
[22] RAO Z P, LU G H, MAHMOOD A,Deactivation and activation mechanism of TiO2and rGO/Er3+-TiO2during flowing gaseous VOCs photodegradation., 2021, 284: 119813.
[23] WANG C Y, RAO Z P, MAHMOOD A,Improved photocatalytic oxidation performance of gaseous acetaldehyde by ternary g-C3N4/Ag-TiO2composites under visible light., 2021, 602: 699-711.
[24] FENG J J, ZHANG D K, ZHOU H P,Coupling P nanostructures with P-doped g-C3N4as efficient visible light photocatalysts for H2evolution and RhB degradation., 2018, 6(5): 6342–6349.
[25] REN H T, JIA S Y, ZOU J J,A facile preparation of Ag2O/P25 photocatalyst for selective reduction of nitrate., 2015, 176–177: 53–61.
[26] SINGH M, ZHOU N J, DILIP K. PAUL,IR spectral evidence of aldol condensation: acetaldehyde adsorption over TiO2surface., 2008, 260(2): 371–379.
[27] XU B Y, ZHU T, TANG X Y,Heterogeneous reaction of formaldehyde on the surface of TiO2particles., 2010, 53: 2644–2651.
[28] BATAULT F, THEVENET F, HEQUET V,Acetaldehyde and acetic acid adsorption on TiO2under dry and humid conditions., 2015, 264: 197–210.
[29] RASKO´ J, KISS J. Adsorption and surface reactions of acetaldehyde on TiO2, CeO2and Al2O3., 2005, 287(2): 252–260.
[30] BIRGER H, DIETER T, SAMMY V,Elucidating the photocatalytic degradation pathway of acetaldehyde: an FTIRstudy under atmospheric conditions., 2011, 106(3/4): 630–638.
[31] ZHANG W P, LI G Y, LIU H L,Photocatalytic degradation mechanism of gaseous styrene over Au/TiO2@CNTs: relevance of superficial state with deactivation mechanism., 2020, 272: 118969.
1994~1997年, 我师从郭景坤先生在中国科学院上海硅酸盐研究所攻读博士学位, 先生时任所长, 工作非常繁忙。尽管如此, 每逢节假日, 先生都会邀请我们去他家里做客, 一来改善我们伙食, 二来也是在轻松的氛围中了解我们的工作和生活, 他也乐于和我们分享他的学习与工作经历, 引导我们不怕困难, 永葆乐观向上。先生后来生病, 讲话不太方便了, 他就重拾以前的爱好—画油画、拉小提琴, 每次我们去家中看他, 他都会兴致勃勃地向我们展示他的画作, 让我们聆听他的琴声……在与先生近30年的相处时光中, 他待学生们永远是和蔼可亲、鼓励有加, 让我们如沐春风; 遇到什么困难, 他总是乐观面对, 让我们感受着他的坚韧和力量。每每回想起来这些, 我们的心里总是暖暖的。

左:孙静 右:郭景坤先生
(孙静)
Supporting materials
Two-dimensional g-C3N4Compositing with Ag-TiO2as Deactivation Resistant Photocatalyst for Degradation of Gaseous Acetaldehyde
XUE Hongyun1,2, WANG Congyu1, MAHMOOD Asad1, YU Jiajun1,2, WANG Yan1, XIE Xiaofeng1, SUN Jing1
(1. State Key Latoratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China; 2. University of Chinese Academy of Sciences, Beijing 100049, China)
Table S1 Assignment of FT-IR bands observed for AT sample in the process of dark adsorption and photocatalytic degradation for acetaldehyde

ProductWavenumber/cm–1Mode of vibration 11072β(CH3) 21128, 1166v(C–C) 31315δ(CH3) 41339δas(CH3) 51377vs(COO) 61419δs(CH3) 71458, 1473δ(CH2) 81541, 1558vas(COO) 91603v(C=C) 101732, 1716, 1699, 1650v(C=O)
Table S2 Assignment of FT-IR bands observed for CAT sample in the process of dark adsorption and photocatalytic degradation for acetaldehyde
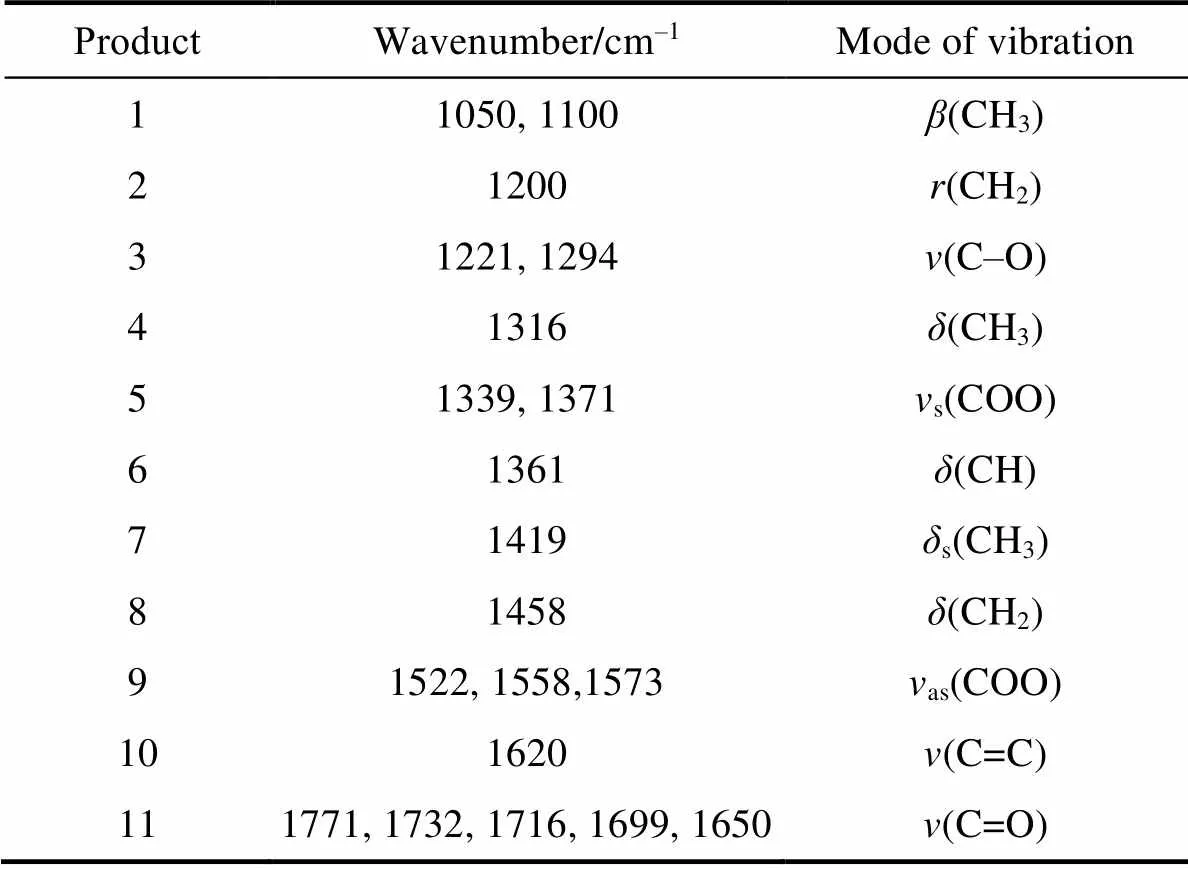
ProductWavenumber/cm–1Mode of vibration 11050, 1100β(CH3) 21200r(CH2) 31221, 1294v(C–O) 41316δ(CH3) 51339, 1371vs(COO) 61361δ(CH) 71419δs(CH3) 81458δ(CH2) 91522, 1558,1573vas(COO) 101620v(C=C) 111771, 1732, 1716, 1699, 1650v(C=O)
二维g-C3N4与Ag-TiO2复合光催化剂降解气态乙醛抗失活研究
薛虹云1,2, 王聪宇1, MAHMOOD Asad1, 于佳君1,2, 王焱1, 谢晓峰1, 孙静1
(1. 中国科学院 上海硅酸盐研究所 高性能陶瓷与超细微结构国家重点实验室, 上海 200050; 2. 中国科学院大学, 北京 100049)
光催化剂失活是影响其在去除低浓度VOCs的实际应用中的主要因素之一。本研究将TiO2与2D石墨碳氮化碳(g-C3N4)复合, 显著提高了光催化剂的稳定性。当Ag改性的Ag-TiO2(AT)用于降解乙醛气体时, 反应60 min后开始发生失活现象, 反应延长至400 min则完全失活。而AT与g-C3N4复合改性后的样品g-C3N4/ Ag-TiO2(CAT)具有优异的光催化性能和稳定性, 反应至600 min未发生失活。原位FT-IR、PL和光电流的研究表明, 当AT催化降解乙醛时, 反应中间体会在表面积累导致催化剂失活。而引入的g-C3N4可以为中间体提供更多的吸附位点, 从而提高稳定性。此外, 引入g-C3N4还有利于电荷分离和产生活性氧物种, 促进乙醛和中间体降解。本研究揭示了2D材料在开发稳定可持续降解VOCs的光催化剂方面的实用性。
光催化; 二氧化钛; g-C3N4; 抗失活; 原位漫反射红外光谱
TQ174
A
1000-324X(2022)08-0865-08
10.15541/jim20220123
date:2022-03-07;
date: 2022-04-05;
2022-04-26
Key Collaborative Research Program of the Alliance of International Science Organizations (ANSO-CR-KP-2020-13); National Key Research and Development Program of China (2021YFE0110400); National Natural Science Foundation of China (41907303, 52072387); Shanghai Commission of Science and Technology Program (19DZ1202600, 20DZ1204100); State Key Laboratory Director Fund of SICCAS (Y9ZC0102)
XUE Hongyun (1998–), female, Master candidate. E-mail: hyxue0310@163.com
薛虹云(1998–), 女, 硕士研究生. E-mail: hyxue0310@163.com
MAHMOOD Asad. E-mail: amkhan036@yahoo.com; SUN Jing, professor. jingsun@mail.sic.ac.cn.
MAHMOOD Asad. E-mail: amkhan036@yahoo.com; 孙静, 研究员. E-mail: jingsun@mail.sic.ac.cn.
