毛细管电色谱-电喷雾电离-飞行时间质谱分离分析盐酸地尔硫卓和盐酸维拉帕米混合手性药物
2021-07-14唐艺旻李英杰高立娣秦世丽靳凤龙刘树仁
唐艺旻, 李英杰, 高立娣, 秦世丽*, 靳凤龙, 刘树仁
( 1.齐齐哈尔大学化学与化学工程学院,黑龙江齐齐哈尔 161006;2.齐齐哈尔食品药品检验检测中心,黑龙江齐齐哈尔 161005;3.浙江大学环境与资源学院环境健康研究所,浙江杭州 310012)
盐酸地尔硫卓和盐酸维拉帕米同为钙离子拮抗剂[1,2],可用于治疗高血压,心绞痛等疾病,其结构如图1所示。这两种药物的药理药效极其相似,在临床用药过程中,常常交换使用,但是盐酸维拉帕米对冠状动脉痉挛的预防效果仅为盐酸地尔硫卓的1/8~1/3。值得关注的是这两种药均为手性药物且以外消旋体供药,这会导致药效的降低甚至危害人体健康[3],但有关这两种药物的手性拆分方法却鲜见报道。因此,探索这两种混合药物的分离检测方法无论对临床治疗还是在基础研究方面都具有重要意义。

图1 盐酸地尔硫卓(A)和盐酸维拉帕米(B)结构式Fig.1 Structures of diltiazem hydrochloride(A) and verapamil hydrochloride(B)
在过去的几十年中,基于高效液相色谱(HPLC)[4,5]、气相色谱(GC)[6]、超临界流体色谱(SFC)[7]、薄层色谱(TLC)[8]、毛细管电泳(CE)[9,10]和毛细管电色谱(CEC)[11]等方法的对映体拆分技术均得到不同程度的发展。CEC具备HPLC的高效性和CE的高选择性,其与β-环糊精[12]或其衍生物的结合,改善了手性药物在CEC中的分离效果。Zou等[13]通过将PDA/S-β-CD附着到涂覆有Au NPs的毛细管上制成开管柱,实现10种手性药物的基线分离。CEC-MS法[14]在CEC的基础上增加了具有定性分析功能的MS,这使得分析范围更广,灵敏度更高。Yan等[15]以HP-β-CD为手性选择剂,应用CEC-MS法同时对延胡索提取物中的四氢巴马汀和卡丹汀进行对映选择性分析,该方法无需任何预纯化即可直接用于测定复杂基质中的分析物。本文采用实验室自制的顺丁烯二酸酐-β-环糊精(MAH-β-CD)[16]作为手性固定相,制备MAH-β-CD电色谱整体柱,采用毛细管电色谱-电喷雾电离-飞行时间质谱(CEC-ESI-TOF MS)对盐酸地尔硫卓和盐酸维拉帕米混合手性药物进行分离检测,获得结果满意。
1 实验部分
1.1 仪器与试剂
7100 CE毛细管电泳仪,6230 飞行时间质谱仪(美国,安捷伦公司);S-4300扫描电子显微镜(日本,日立公司)。
手性药物标准品(纯度99%,中国药品生物制品检定所);甲基丙烯酸-三甲基硅烷丙酯(γ-MAPS)、聚甲基丙烯酸缩水甘油酯(GMA)、乙二醇二甲基丙烯酸酯(EDMA)(分析纯,阿拉丁试剂);偶氮二异丁腈(AIBN)、2-丙烯酰胺基-2-甲基-1-丙磺酸(AMPS)(美国Fluka公司)。实验用水均为超纯水。
盐酸地尔硫卓片剂、盐酸维拉帕米片剂(中国天津田边制药有限公司)。
1.2 整体柱制备
将硅烷化试剂(γ-MAPS∶丙酮=1∶1)注入毛细管柱中后,将两端封口。室温放置24 h,甲醇冲洗,用N2吹干,待用。称量0.15 g GMA,0.10 g EDMA,0.10 g 环己醇,0.24 g 正十二醇,0.0018 g AIBN,0.0016 g AMPS 和0.10 g MAH-β-CD的DMSO溶液(0.2 g/mL),超声混匀。将混合物填充至毛细管柱中24.0 cm处(柱总长130 cm),40 ℃反应12 h后,备用。
1.3 样品的制备
取一片盐酸地尔硫卓或盐酸维拉帕米片剂重量的粉末,溶于10 mL甲醇中,超声溶解,6 000 r/min离心10 min,上清液用甲醇稀释至适当浓度,每次用前过0.22 μm滤膜。
1.4 CEC分离条件
采用pH=4.50的20 mmol/L NH4Ac作为运行缓冲溶液,运行电压20 kV,分离温度20 ℃,压力进样(5 kPa×3 s),波长200 nm处紫外检测。
1.5 ESI -TOF/MS 检测条件
离子源:电喷雾电离,正离子模式;扫描范围:m/z300~500,雾化气压力241 kPa,干燥气为N2(纯度99.999%):温度300 ℃,流速8 L/min;鞘流液为50%甲醇,流速为0.3 mL/min。
2 结果与讨论
2.1 电色谱整体柱扫描电镜表征
电色谱整体柱扫描电镜(SEM)图如图2。可见聚合物成功键合到柱内壁上,柱内形成了致密的网状结构,其形态错综复杂,由此推测该柱有较强的机械强度。而且柱内较大的孔径保证了一定的通透性。

图2 手性柱内固定相扫描电镜(SEM)图Fig.2 SEM images of the stationary phase in monolithic column A.cross-section;B.a part of cross-section.
2.2 CEC分离条件优化
2.2.1 运行缓冲溶液种类及浓度的选择本实验考虑到避免MS中雾化器堵塞,以及离子源污染进而影响检测,损坏仪器等问题,故选择易挥发的NH4Ac为CEC-MS的运行缓冲溶液。在一定浓度范围内,离子强度和电渗流均随缓冲溶液浓度的增加而增加,但浓度过大产生气泡,不利于对映体分离。本实验选择20 mmol/L NH4Ac为缓冲体系的浓度。
2.2.2 缓冲溶液pH值的选择毛细管表面电荷密度和电渗流平衡受缓冲溶液pH的直接影响,进而影响组分分离。由于盐酸地尔硫卓有2个手性中心,故其有4个异构体;盐酸维拉帕米有1个手性中心,其含有2个异构体。而在本实验所考察的pH范围内,两混合药物对映体6组分中有4组分被检测出来,且实现不同程度的分离(混合手性药物对映体的4组分峰分别用A、B、C、D表示),4组分两两间在不同pH值下的分离度(Rs)见表1。结果表明,pH=4.50时4组分的分离效果较好,故最佳pH值选为4.50。

表1 pH对两种混合手性药物分离的影响
Rs1:Resolution of peak A and B;Rs2:Resolution of peak B and C;Rs3:Resolution of peak C and D.
2.2.3 运行电压的选择分析物迁移时间随着电压增大而减小,分离度则是先增大后减小。分离度减小是由于过大的电压使得柱内产生气泡,进而影响组分分离。图3为不同电压下混合手性药物盐酸地尔硫卓和盐酸维拉帕米电色谱分离图,电压为20 kV时,两混合手性药物各组分间分离度达最大,从而确定运行电压为20 kV。

图3 电压对两种混合手性药物分离的影响Fig.3 Effect of voltage on separation of two chiral drugs mixtureConditions:20 mmol/L NH4Ac(pH=4.50);column temperature:20 ℃;detection wavelength:200 nm.
2.3 ESI-TOF/MS检测条件的优化
2.3.1 鞘流液的选择分析物的电离强度,背景信号值等很大程度上受鞘流液的种类和浓度的影响。与异丙醇相比,甲醇为鞘流液时背景噪音较低且分析物电离强度较大。当甲醇含量20%~60%时,分析物电离强度各有不同,结果如图4。鞘流液中甲醇含量为50%时,混合手性药物药物的4组分(A、B、C、D)电离强度均达到最大值,故浓度为50%的甲醇作为本实验的鞘流液。
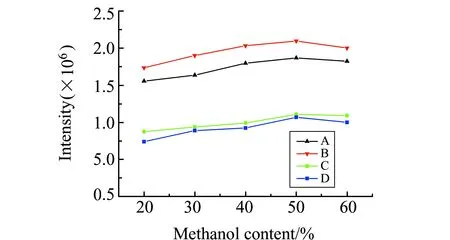
图4 鞘流液中甲醇含量对电离的影响Fig.4 Effect of methanol content on the ionization of the sheath fluid
2.3.2 鞘流液流速的选择考察流速范围:0.2~1.0 mL/min,当流速由0.2 mL/min增至0.4 mL/min时,分析物电离强度略微增大;流速大于0.4 mL/min时,稀释作用导致电离强度减小。故选择0.3 mL/min作为鞘流液流速。
2.4 混合手性药物的CEC-TOF MS定性分析
在本实验优化的条件下,检测出两混合手性药物的4组分。图5为混合手性药物总离子流图色谱图。为了验证检测出两混合手性药物4组分的主要成分,采用MS对图5中4组分峰进行定性分析,各组分提取质谱图见图6。

图5 盐酸地尔硫卓和盐酸维拉帕米混合物的总离子流(TIC)色谱图Fig.5 TIC chromatogram of diltiazem hydrochloride and verapamil hydrochloride mixtureConditions:20 mmol/L NH4Ac(pH=4.50);separation voltage:20 kV;column temperature:20 ℃;detection wavelength:200 nm.
图6A、6B分别为保留时间16.41 min和16.54 min组分的质谱图,其中m/z为 415.04840和415.16501的离子峰均为盐酸地尔硫卓的[M-HCl+H]+峰。图6A中,其失去15(CH3)和32(H3CO)的碎片后分别产生m/z为400.02151 和383.88901的质谱峰;图6B中,其失去15(CH3)和32(H3CO) 碎片后分别产生m/z为 400.23206和383.18006的质谱峰,由此推断,图6A、6 B为盐酸地尔硫卓异构体。图6C、6D分别为保留时间17.61 min和17.91 min组分的质谱图,其中m/z455.28536和 455.28555的离子峰均为盐酸维拉帕米的[M-HCl+H]+峰。图6C中,其失去15(CH3)、32(H3CO)和44(CH(CH3)2)的碎片后分别产生m/z为 440.74425、423.96664和411.70574的质谱峰;图6D中,其失去15(CH3)、32(H3CO)和44(CH(CH3)2)的碎片后分别产生m/z为 440.26876、423.27350和411.15385的质谱峰,由此推断,图6C、6D为盐酸维拉帕米的对映异构体。

图6 四个组分的质谱图Fig.6 MS spectra of four componentsA,B:diltiazem hydrochloride isomer;C,D:verapamil hydrochloride isomer.
2.5 实际样品的分离定性
为检验整体柱的实际应用价值,使用该柱在“1.4”和“1.5”的最佳CEC及MS条件下,分离分析市售盐酸维拉帕米和地尔硫卓混合手性药物。如图7所示,检测出的4组分达到了基线分离,说明市售混合手性药物被初步分离检测。

图7 混合实际药品总TIC色谱图Fig.7 TIC chromatogram of a mixture consisting of actual diltiazem hydrochloride and verapamil hydrochloride
3 结论
本文以MAH-β-CD整体柱为色谱分离柱,利用CEC-ESI-TOF MS联用技术对盐酸地尔硫卓和维拉帕米混合手性药物进行分离分析。在最佳条件下,混合药物的6组分中有4组分达到基线分离并被检测出来。由于盐酸地尔硫卓含有2个手性中心,采用本实验方法并没有实现该药物对映体的完全分离。但该方法可同时成功检测出盐酸地尔硫卓的2种异构体及盐酸维拉帕米的2种异构体,为今后混合手性药物的检测及拆分奠定了一定的基础。
