薄荷基β-二酮铁的合成及其在偶联反应中的应用研究
2021-06-07张令君彭天英贺国文
张 劲,张令君,彭天英,贺国文
(湖南城市学院 材料与化学工程学院,湖南 益阳 413000)
金属催化剂具有干净、高效、选择性强的特点,特别是廉价环保、可以回收利用的金属催化剂具备广阔的应用前景.然而,金属配合物的中心金属离子一般是重金属或者稀有金属,它们所具有的毒性和昂贵的价格严重阻碍了其在实际生产环境中的推广应用[1-3].在能源与环境问题日益严重的今天,继续使用高毒性、来源匮乏的贵金属来制备催化剂是不符合潮流的.因此,开发廉价、环保低毒、来源广泛的金属催化剂具有非常重要的意义[4].
铁在自然界分布极为广泛,是地壳中含量位居第二的金属元素.铁催化剂具有环境友好性强、来源广泛、价格低廉等特点.目前,在CO2的电化学还原[5-6]、化学键活化[7]、脱羰基功能化[8]等方面铁作为催化剂的研究报道很多,但与其它过渡金属相比,在不对称催化反应中铁催化剂的研究仍然相对较少.也有研究指出,铁催化剂的催化活性中心并非铁离子,而是其中所混杂的铜盐离子.早在1971年,Kochi等就已经发现铁盐可被用来催化Grignard试剂与烯基和芳基卤化物的交叉偶联[9].
当用铁催化溴代乙烯基苯基硫醚底物与i-PrMgCl反应时,偶联所得产物是具有立体专一性的.磺酰基烯烃和Grignard试剂也可以发生偶联反应,但是得到的产物常常是顺反异构体的混合物.在有些情况下,某些反应还是可以得到高产率和选择性比较好的产物的.但需要注意的是在n-BuLi存在的情况下,使用同样的催化剂,磺酰基可以自偶联生产烯烃化合物.Grignard试剂也可以和酰卤在足够催化量的铁化合物下发生偶联反应,高产率地生成酮[10].
在不对称催化反应中,研究人员往往更多地关注反应产率和对映选择性,从而忽略了反应过程的可操作性、经济成本、绿色安全等现在看来非常重要的因素.通常,不对称反应所用催化剂的金属中心都具有毒性,且其价格昂贵、来源匮乏.因此,手性催化剂中心金属的选择是相当重要的.相对于手性钌、铑、铱等传统催化体系而言,手性铁催化剂的研究才刚刚起步.铁离子具有安全无毒、成本低廉、来源广泛等优点,随着对其研究的增多,铁催化剂在不对称催化反应中的应用范围将进一步拓宽,其催化活性和对映选择性也会进一步提高.相信在不久的将来,更多优秀、环保的铁催化剂将会被研究、开发并应用于更多类型的不对称催化反应[11-12].
本文从薄荷醇出发,使其经卤化反应、格氏反应、酰氯化反应得到薄荷基甲酰氯,再与甲基锂反应生成薄荷基β-二酮,并在甲醇钠的作用下与三氯化铁反应得到薄荷基β-二酮铁配合物.通过核磁共振和红外光谱对部分中间产物和最终产物进行表征,并将薄荷基β-二酮铁配合物作为催化剂,加入苯基溴化镁和2-溴丁烷的偶联反应,考察其在偶联反应中的催化效果.
1 实验部分
1.1 主要仪器与试剂
核磁共振仪(NMR,BRUKER AV-400,溶剂CDC13,内标TMS);气质联用仪(AGILENT 5975 GC);质谱仪(AB SCIEX TRIPLETOF 4600).主要试剂包括薄荷醇、三氯化铁、甲基锂.
1.2 薄荷基氯的合成
薄荷基氯的合成原理[13]如图1所示.在1 000 mL的三口瓶中加入浓盐酸200 mL,搅拌,冰水下冷却,加入536.00 g(3.94 mol)无水氯化锌,待完全溶解后撤去冰水浴,一次性加入薄荷醇198.00 g(1.27 mol);在35 ℃下反应5 h后静置分层约1 h,分出有机相,水相用石油醚萃取(3×50 mL);合并有机相,有机相水洗(3×50 mL),再用浓硫酸洗至浓硫酸无色,有机相再水洗至中性,无水硫酸钠干燥过夜;过滤,蒸除滤液中溶剂,剩余物减压蒸馏得薄荷基氯204.02 g,收率92%(收集90~92 ℃/1.33 kPa的馏分).
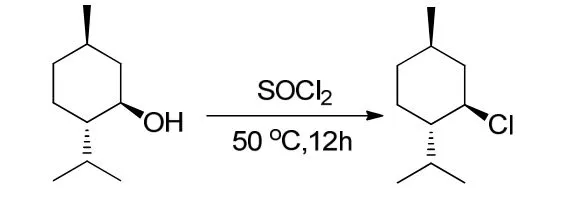
图1 薄荷基氯的合成
1H NMR(400 MHz, CDCl3, TMS, δ): 0.76(d,J=6.4 Hz, 3 H), 0.89(d,J=6.3 Hz, 6 H), 0.92(m, 2 H), 1.4(m, 2 H), 1.65(m, 2 H), 1.80(m, 1 H), 2.19(m,1 H), 2.35(m, 1 H), 3.78(dt,J1=11.0 Hz,J2=4.2 Hz,1 H).
13C NMR(100 MHz, CDCl3, TMS, δ):15.1(CH3), 21.0(CH3), 21.9(CH3), 24.2(CH2),27.1(CH), 33.3(CH), 34.2(CH2), 46.7(CH2),50.4(CH), 63.8(CH).
1.3 薄荷基甲酸的合成
将7.20 g(0.30 mol)镁粉加入到500 mL的三口瓶中,装上机械搅拌器、恒压滴液漏斗和回流冷凝管,反应体系用氮气保护;向反应瓶加入40 mL无水THF和几粒碘单质,加热回流后加入约0.2 mL溴乙烷引发;待碘的黄色退去后搅拌,向滴液漏斗中慢慢滴加薄荷基氯(50 mL,0.25 mol)的THF(160 mL)溶液,约2 h滴完;滴完后,继续加热回流搅拌,反应8 h使其反应完全,冷却;反应体系转移至低温反应浴中,除去恒压滴液漏斗和回流冷凝管,装上干燥管和通气管;待反应体系降温至−80 ℃后,通入二氧化碳气体,每秒约1~2个气泡,鼓泡15 h后停止反应;随即把反应液倒入60 mL的6 mol/L盐酸中搅拌,会伴有大量气泡产生;分层后分出有机相,水相用乙醚萃取(3×40 mL);合并有机相,有机相除去溶剂后水洗(3×20 mL),用5%的NaOH溶液萃取(5×30 mL);水相用乙醚洗涤2次(2×50 mL)后,用盐酸酸化到pH=3左右,当有大量白色浑浊出现时静置分层,分出有机相,水相用乙醚萃取(4×50 mL);合并有机相,有机相水洗(3×20 mL),用无水硫酸钠干燥过夜;除去溶剂,结晶得白色晶体18.50 g,收率40.0%,m.p. (熔点) =69.7~70.1 ℃.薄荷基甲酸的合成原理[13]如图2所示.H), 0.9~1.1(m, 2 H), 1.21(q,J=12.2 Hz, 1 H), 1.35(m,1 H, -CHMe2), 1.50(1 H,J1=3.0 Hz,J2=11.6 Hz),1.66~1.77(m, 3 H), 1.90~1.94(m, 1 H), 2.32(dt,J1=11.7 Hz,J2=3.21 Hz, 1 H, -CHCOOH-).
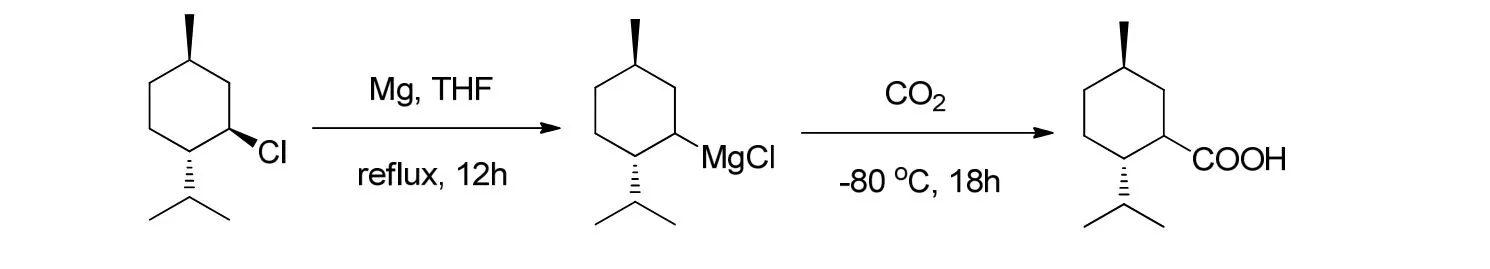
图2 薄荷基甲酸的合成
13C NMR(100 MHz, CDCl3, TMS, δ):16.0(CH3), 21.3(CH3), 22.3(CH3), 23.8(CH2),29.3(CH), 32.0(CH), 34.5(CH2), 38.8(CH2),44.2(CH), 47.7(CH), 183.0(CO2H).
1.4 薄荷基甲酰氯的合成
薄荷基甲酰氯的合成[13]如图3所示.
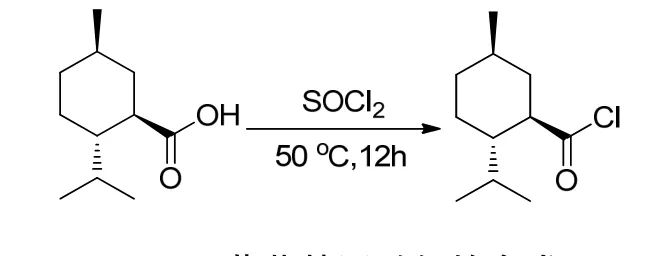
图3 薄荷基甲酰氯的合成
取薄荷基甲酸18.40 g(100 mmol),加入100 mL圆底烧瓶中,然后加入44 mL(600 mmol)氯化亚砜搅拌,使固体完全溶解;装上冷凝管和干燥管,尾气用10%的NaOH溶液吸收,加热回流,搅拌反应4.5 h后蒸去多余的二氯亚砜;剩余液减压蒸馏,可得无色液体16.80 g(84 mmol),收率83.2%(收集164~168 ℃/2.66 kPa的馏分).
1H NMR(400 MHz, CDCl3, TMS, δ):0.81~2.07(m, 18 H, menthyl H), 3.34(dt,J1=11.7 Hz,J2=3.21 Hz, 1 H, -CHCOCl).
13C NMR(100 MHz, CDCl3, TMS, δ):15.8(CH3), 21.4(CH3), 22.1(CH3), 23.5(CH2),29.2(CH), 32.0(CH), 34.8(CH2), 38.3(CH2),44.8(CH), 59.2(CH), 176.8(CO2H).
1.5 薄荷基β-二酮的合成
薄荷基β-二酮的合成[13]如图4所示.
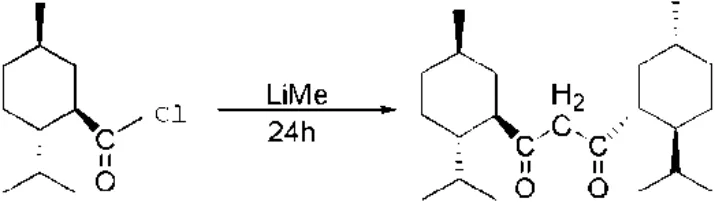
图4 薄荷基β-二酮的合成
在氩气保护下,往装有恒压滴液漏斗的25 mL两口瓶中加入3.60 mL(5.50 mmol)甲基锂的乙醚溶液(1.6 mol/L)和10 mL无水乙醚;再经恒压滴液漏斗在0 ℃下缓慢滴加1.01 g(5.00 mmol)薄荷基甲酰氯和上述相同并等量的反应溶剂组成的溶液至磁力搅拌的两口瓶中;滴加完毕后,在上述相同的温度下反应24 h,再用冰水淬灭反应;收集有机层,用5 mL的乙醚萃取水层后合并有机相,有机相用无水硫酸钠干燥后除去溶剂,将剩余油状物进行柱层析(固定相200~300目硅胶,洗脱剂V二氯甲烷/V石油醚=1∶20)分离后得橙黄色固体;用95%无水乙醇对其进行重结晶得白色产物薄荷基β-二酮,产率为67%.
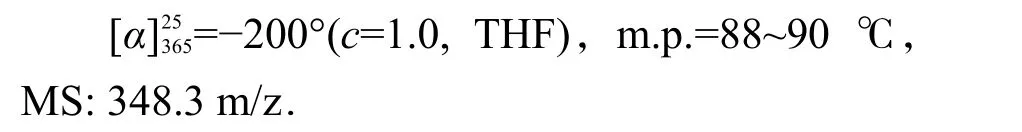
1.6 薄荷基β-二酮铁配合物的合成
薄荷基β-二酮铁配合物合成如图5所示.
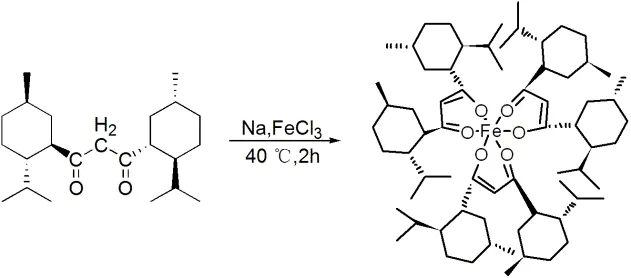
图5 薄荷基β-二酮铁配合物的合成
在氮气保护下,向装有温度计和滴液漏斗的三口瓶中加入0.6 g金属钠和30 mL绝对无水甲醇,待金属钠完全反应后,加入8 mmol薄荷基β-二酮,在40 ℃下搅拌反应0.5 h;将FeCl3·6H2O(0.8 g, 2.5 mmol)溶在10 mL甲醇中,滤去不溶物,滤液移至滴液漏斗,并滴至上述反应混合物中,立即有红色沉淀产生;滴完后,在40 ℃下搅拌反应2 h,冰水浴冷却,过滤得到红色固体;用甲醇洗涤固体2次,抽滤,得红色粗产物;将粗产物晾干后溶在10 mL正己烷中,过滤,除去不溶性杂质,蒸去正己烷,得红棕色固体,收率40%.
2 结果与讨论
2.1 薄荷基β-二酮铁配合物红外解析
薄荷基β-二酮铁配合物的红外光谱如图6所示.从图6可发现,配合物中存在-CH3(2 953.1,2 869.6和1 385.0 cm−1)、C-C(1 422.5 cm−1)、C-O(1 558.9 cm−1)、C-CH3(1 265.4 cm−1)等基团,这证明了薄荷基β-二酮负离子的存在.薄荷基β-二酮负离子与Fe3+配位后,形成了3个大π键,环上电子云向中心离子方向移动,使C=O键减弱.这与典型的羰基位置不一样,其伸缩振动吸收峰移向低频,在红外谱上表现出1 573 cm−1的强吸收峰.红外谱中的780,675和484 cm−1等峰,可以被归属为Fe-O键的伸缩振动和螯合环的变形振动[14-15],这说明薄荷基β-二酮的O与Fe3+形成了配位键并已成环.由此可知,薄荷基β-二酮铁配合物被成功合成.
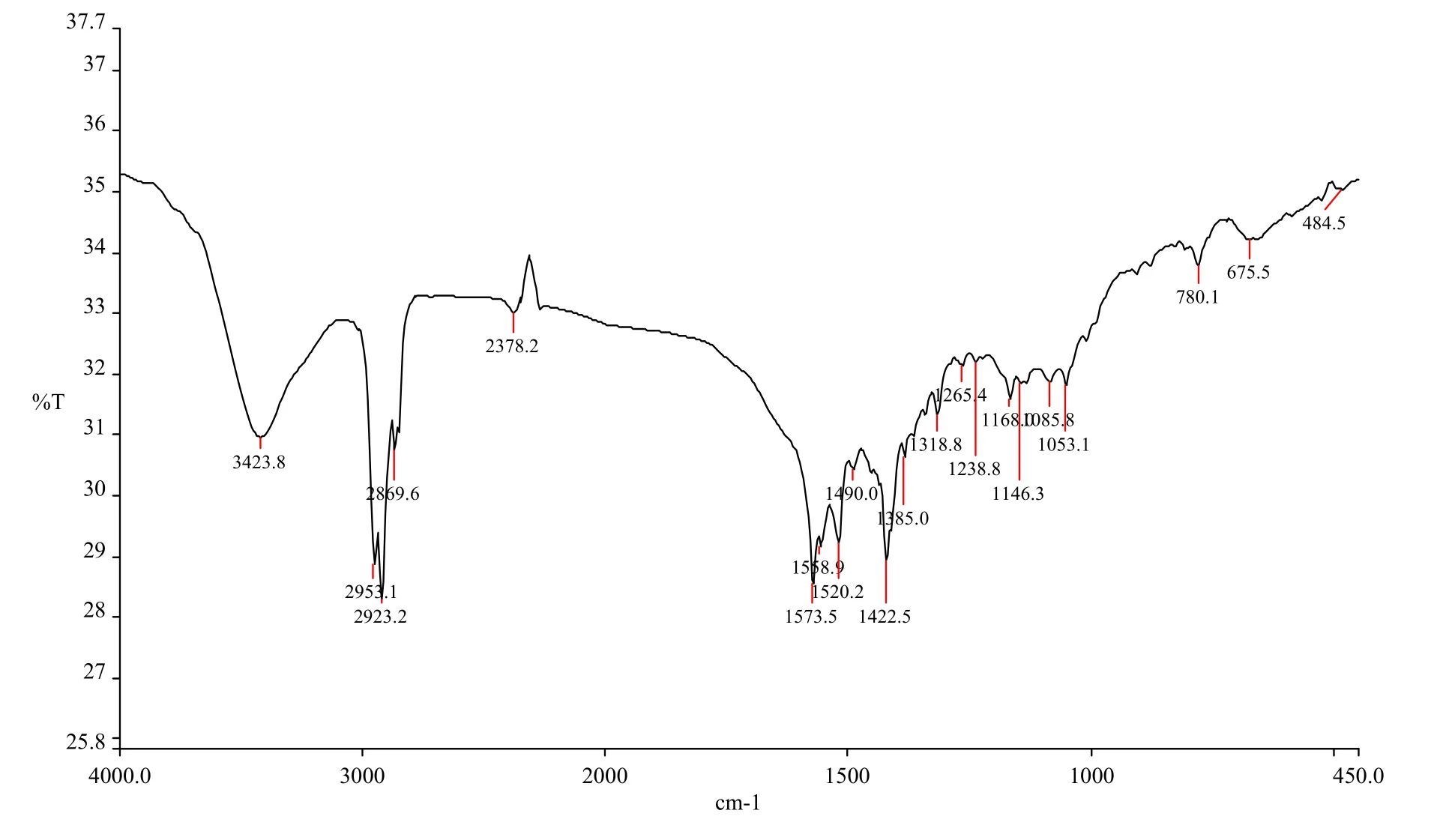
图6 薄荷基β-二酮铁配合物的红外光谱
2.2 薄荷基β-二酮铁配合物对偶联反应的催化性能影响
在0 ℃下,向装有恒压滴液漏斗和氮气保护装置的50 mL三口瓶中加入0.05 g薄荷基β-二酮铁配合物、0.11 mL 2-溴丁烷、5 mL THF,搅拌30 min后,滴加1 mL的苯基溴化镁(1 mol/L THF);滴加完毕,继续搅拌一段时间,再向反应体系中加入10 mL饱和氯化铵溶液,搅拌5 min;用乙醚萃取产物,再用无水硫酸钠干燥过夜,过滤以除去不溶性杂质;蒸去乙醚,得无色透明油状液体,对其进行气质检测.苯基溴化镁与正溴丁烷的偶联反应如图7所示,薄荷基β-二酮铁配合物在偶联反应中的催化效果如表1所示.

图7 苯基溴化镁与正溴丁烷的偶联反应
由表1可知,薄荷基β-二酮铁配合物在偶联反应中的催化效果并不明显,延长反应时间对结果也没有影响.其原因是薄荷基β-二酮配体的空间位阻过大,阻碍了活性中心与反应底物的接触,导致反应活性降低,目标产物生成少.

表1 薄荷基β-二酮铁配合物对偶联反应的催化影响
3 结论
本文研究了薄荷基β-二酮铁配合物的合成,对产物及部分中间体的结构用NMR和IR进行了表征,相关图谱数据也证明合成了这些物质.同时,研究还发现,使用薄荷基β-二酮铁配合物作为苯基溴化镁和2-溴丁烷的偶联反应催化剂,其催化效果并不明显.后期研究应致力于寻找匹配该类催化剂的体系,发挥其催化效果.
