玉米穗轴粗全基因组关联分析
2021-04-27曹言勇李会勇
马 娟 曹言勇 李会勇
玉米穗轴粗全基因组关联分析
马 娟 曹言勇 李会勇*
河南省农业科学院粮食作物研究所, 河南郑州 450002
玉米穗轴粗是一个影响玉米产量和穗轴产量的重要性状, 其遗传机制的解析可以对高产育种提供指导。本研究以309份玉米自交系为材料, 利用测序基因型分型技术对其进行基因型鉴定。采用FarmCPU (fixed and random model circulating probability unification)、MLMM (multiple loci mixed linear model)和CMLM (compressed mixed linear model)方法, 对2017年和2019年河南原阳、河南郸城、河南虞城、海南三亚以及最佳线性无偏估计值环境的穗轴粗, 进行全基因组关联分析, 共鉴定12个与穗轴粗显著关联的SNP (single nucleotide polymorphisms) (<8.60E-07)。其中, S4_29277313利用FarmCPU和MLMM方法在2017年原阳均检测到。S1_29006330、S2_170889116、S2_2046026464和S4_83821463的表型变异解释率介于10.23%~14.17%, 为主效SNP。而且, S1_29006330位于已定位的穗轴粗QTL (quantitative trait loci)区间内。共挖掘17个候选基因, 其中(wall-associated receptor kinase-like 14)、转录因子(zinc-finger protein expressed in inflorescence meristem 35)、(HMG-Y-related protein A)、组蛋白-赖氨酸N甲基转移酶(trithorax 4)和(xyloglucan endotransglucosylase/hydrolase protein 32), 可能是影响穗轴粗的重要基因。挖掘的4个穗轴粗主效SNP和5个候选基因可以为分子标记辅助育种、精细定位和基因克隆提供信息。
玉米; 全基因组关联分析; FarmCPU; 穗轴粗
玉米穗轴粗是一个重要的穗部性状, 与玉米产量相关性状具有密切关系。穗轴粗与穗粗具有显著正相关关系, 相关系数高达0.57~0.77[1-4], 与穗行数表现为中度极显著正相关(= 0.41~0.45,<0.01)[1-2,5]。此外, 穗轴粗与单穗粒重(= 0.21~0.39)[2-3,6]、穗长(= 0.34~0.36)[1,4]、行粒数(= 0.25)[1]、单穗重(= 0.21~0.22)[1,4]、粒长(= 0.27)[4]、粒宽(= 0.17~0.23)[4]和粒厚(= 0.20)[2]也表现出显著正相关。玉米穗轴同时作为重要的工业原材料, 可以用来制备生物燃料纤维素乙醇[7], 在可持续能源发展中具有重要作用。穗轴粗是影响穗轴的一个重要性状, 与穗轴重存在较高正相关关系(= 0.58~ 0.61,<0.01)[1-2]。因此, 解析玉米穗轴粗的遗传机制, 对玉米产量和穗轴产量的提高均具有重要意义。
数量性状定位和全基因组关联分析(genome-wide association study, GWAS)是解析穗轴粗遗传构成的重要方法。Yi等[8]利用RIL (recombinant inbred line)群体和永久F2群体共鉴定13个控制穗轴粗的数量性状位点(quantitative trait loci, QTL), 其解释表型变异率为4.3%~8.7%。Guo等[2]利用郑58×昌7-2构成的231个F2:3家系在2个密度条件下挖掘13个穗轴粗QTL, 其中3个QTL的表型变异解释率为13.65%~24.71%, 为控制穗轴粗的主效QTL。Choe等[1]利用韩国糯玉米构成的F2:3群体鉴定到7个控制穗轴粗的QTL, 解释表型变异率为4.4%~12.9%。王帮太等[9]通过元分析(meta-analysis)整合了产量相关性状QTL, 发现穗轴粗存在5个一致性QTL, 其中, 2个Meta-QTL在籽粒产量的2个区域中出现。Su等[3]利用复合区间作图和最小绝对值收敛和选择算子分别鉴定2个和3个穗轴粗QTL。Zhu等[4]利用全基因组关联分析挖掘到5个穗轴粗显著关联SNP (single nucleotide polymorphisms)和6个候选基因。通过连锁分析和关联分析, Zhang等[10]共鉴定13个穗轴粗QTL和25个穗轴粗显著关联SNP。
目前, 有关玉米穗轴粗全基因组关联分析和候选基因的研究报道较少。本研究利用309份材料构成的关联群体对穗轴粗进行全基因组关联分析, 挖掘玉米穗轴粗显著关联位点和候选基因, 为选育穗轴产量高的玉米新品种提供理论基础。
1 材料与方法
1.1 材料和田间试验设计
选用国内玉米核心种质和黄淮海骨干自交系等309份材料作为关联群体。在2017年夏, 309份材料分别种植在河南新乡原阳(Yuanyang, YY)、河南商丘虞城(Yucheng, YC)、河南周口郸城(Dancheng, DC)和海南三亚(Sanya, SY)。2019年夏种植在原阳。采用随机区组试验设计, 2行区, 2粒播, 行距60 cm, 株距25 cm, 每行15株, 共设3个重复。脱粒后, 测量穗轴粗。
1.2 表型数据统计分析
利用R语言对不同环境穗轴粗进行相关性分析。利用QTL IciMapping v4.0[11]对2017年原阳、郸城、虞城、三亚和2019年原阳进行联合方差分析, 并计算广义遗传力和最佳线性无偏估计值(best linear unbiased estimator, BLUE)。
1.3 基因型鉴定和分析
309份自交系采用GBS (genotyping-by-sequencing)简化测序的方法进行基因型鉴定。测序仪为Illumina HiSeq PE150双端测序。利用BWA软件比对B73参考基因组(ftp://ftp.ensemblgenomes.org/pub/plants/ release-36/fasta/zea_mays/dna/Zea_mays. AGPv4.dna. toplevel.fa.gz)。采用SAMTOOLS软件检测群体SNP。以缺失率小于0.10, 杂合率小于0.10, 最小等位基因频率(minor allele frequency, MAF)大于0.05为筛选条件, 获得58,129个SNP用于后续分析。
1.4 全基因组关联分析
利用2017年原阳、郸城、虞城、三亚、2019年原阳和BLUE环境的穗轴粗进行全基因组关联分析。为了控制假阳性和假阴性, 本研究利用FarmCPU (fixed and random model circulating probability unification)[12]、MLMM (multiple loci mixed linear model)[13]和CMLM (compressed mixed linear model)[14]方法的(群体结构) +(亲缘关系)模型进行全基因组关联分析。群体结构值由Structure v2.3.4计算。其中亚群数为1~8, length of burn-in period为5000, 蒙特卡罗重复个数为50,000, 每个亚群数迭代次数为3。根据Δ的结果, 确定亚群数为2时的值用于关联分析。亲缘关系值由TASSEL v5.0软件的Centered_IBS方法计算。显著临界值设置为=0.05/58129=8.60E-07。FarmCPU和MLMM方法显著位点的表型变异解释率(phenotypic variation explained, PVE)采用线性回归方法计算[15], CMLM方法的PVE由软件给出[13]。利用ANNOVAR对显著关联位点挖掘穗轴粗的候选基因。通过检索maizeGDB, 获得候选基因在不同组织的转录表达数据。
2 结果与分析
2.1 穗轴粗表型数据分析
2017年和2019年不同环境关联群体的轴粗变异范围为1~4 cm。从柱状图的拟合曲线上看, 除了YY2019, 其余4个环境的穗轴粗基本符合正态分布(图1)。多环境穗轴粗的广义遗传率为0.77。相关性分析表明不同环境间均存在显著正相关关系(= 0.15~0.51,<0.05) (图1)。而且, 2017年4个环境的穗轴粗相关系数较高。其中, 2017年郸城和虞城穗轴粗的相关系数最高, 为0.51 (<0.001), SY2017和YY2017间的相关系数为0.50 (<0.001)。2019年原阳的穗轴粗与2017年4个环境的相关系数均较低(= 0.15~0.33)。
综合5个环境的联合方差分析表明, 关联群体基因型间存在极显著差异(表1), 说明材料间穗轴粗存在显著的遗传变异。不同环境间和基因型与环境互作也存在极显著差异(表1)。这说明, 尽管穗轴粗的遗传力较高, 也受到环境因素的影响。
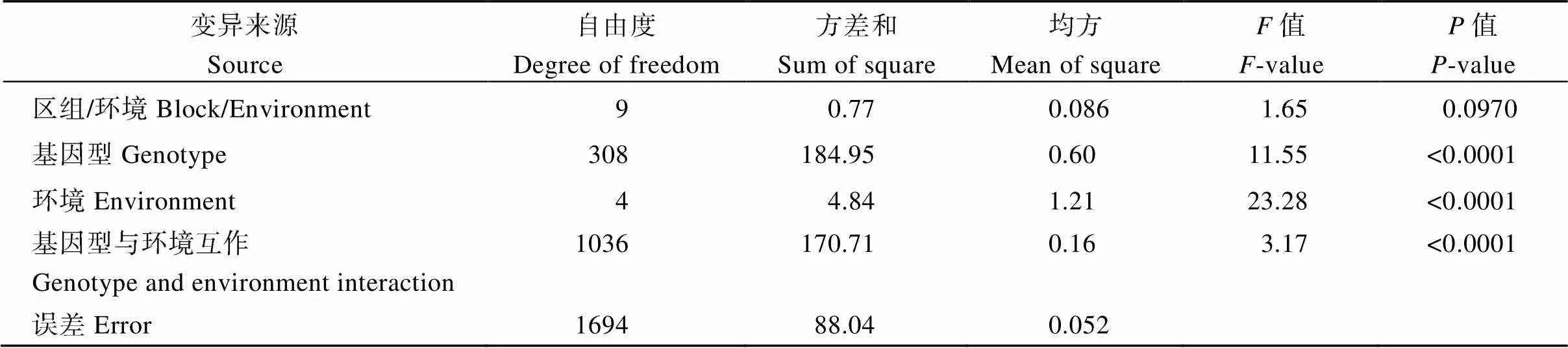
表1 多环境联合方差分析
图中对角线方框为性状的柱状图, 上三角为相关系数和显著性程度。0.05、0.01、0.001 显著水平分别标*、**、***。下三角为不同环境间穗轴粗的散点图。SY、DC、YC和YY分别表示三亚、郸城、虞城和原阳。2017和2019代表年份。
Histograms are showed in diagonal. Correlation values and significant levels represented by asterisk are showed in upper triangular matrix. Significant levels 0.05, 0.01, and 0.001 are denoted by *, **, and ***, respectively. Scatter plots of ear cob diameter are showed in lower triangular matrix. SY, DC, YC, and YY represent Sanya, Dancheng, Yucheng, and Yuanyang, respectively. 2017 and 2019 denote years.
2.2 穗轴粗全基因组关联分析
利用58,129个SNP标记对2017年郸城、虞城、原阳、三亚、2019年原阳和BLUE环境的穗轴粗进行全基因组关联分析, 共检测到12个穗轴粗显著关联SNP, 其中5个位于基因间, 4个位于内含子区和3个位于外显子区(表2)。CMLM方法没有检测到穗轴粗显著关联位点(附图1)。MLMM方法仅在2017年原阳检测到1个显著SNP, 即S4_29277313 (= 7.96E-07), 位于第4号染色体上的外显子区, 解释表型变异小于1%, 为控制穗轴粗的微效位点(图2、表2和附图2)。FarmCPU方法对不同环境(除了2019年原阳)穗轴粗均具有较好的拟合效果(图2和附图2)。该模型共检测到12个显著SNP (= 1.12E-09~6.12E-07) (图2和表2)。其中, BLUE环境检测到4个SNP, 2017年郸城、三亚、虞城和原阳分别检测到2个、2个、1个和3个显著SNP。其中, FarmCPU模型也检测到S4_29277313。FarmCPU模型共检测到4个主效SNP。S1_29006330位于第1号染色体bin1.02, 解释穗轴粗变异率为13.10%。在第2号染色体上, 检测到2个主效SNP, 即S2_170889116和S2_204602646, 分别解释穗轴粗变异率为14.17%和10.23%。S4_83821463位于第4号染色体bin4.05, 解释穗轴粗变异率为12.83%。不同环境间没有检测到相同的SNP, 这也说明穗轴粗受环境的影响较大。
A和B: BLUE环境FarmCPU方法; C和D: 2017年郸城FarmCPU方法; E和F: 2017年三亚FarmCPU方法; G和H: 2017年虞城FarmCPU方法; I和J: 2017年原阳FarmCPU方法; K和L: 2017年原阳MLMM方法。
A and B: FarmCPU in BLUE environment; C and D: FarmCPU in Dancheng in 2017; E and F: FarmCPU in Sanya in 2017; G and H: FarmCPU in Yucheng in 2017; I and J: FarmCPU in Yuanyang in 2017; K and L: MLMM in Yuanyang in 2017.
2.3 穗轴粗候选基因分析
从12个穗轴粗显著关联SNP中共挖掘17个候选基因(表2), 结合maizeGDB中基因的转录表达数据(附图3), 发现(核仁素蛋白)和(HMGA)在穗发育相关的4个组织(16~19 d的分生组织、2~4 mm穗原基、6~8 mm穗原基和雌小穗)中表达量较高, 分别为63.9~157.1和61.5~107.5。编码组蛋白-赖氨素N-甲基转移酶ATX4 (Arabidopsis trithorax 4), 在16~19 d的分生组织、2~4 mm穗原基和6~8 mm穗原基的表达量较高, 达到97.0~119.2。编码的产物是single myb histone 6 (SMH6), 在4个组织中的表达量为15.5~19.3。是ZIM (zinc-finger protein expressed in inflorescence meristem)转录因子ZIM35, 在16~19 d的分生组织中表达量较高(17)。编码染色质组装因子1亚单位FAS1 (chromatin assembly factor 1 subunit FAS1), 在2~4 mm穗原基的表达量最高(21.2)。Zm0000 1d029814编码木葡聚糖内糖基转移酶/脱水酶蛋白XTH32(xyloglucan endotransglucosylase/hydrolase protein 32), 在雌小穗中表达量较高(48), 在16~19 d的分生组织表达量为17.9。候选基因编码细胞壁连接的类受体激酶WAKL14 (wall-associated receptor kinase-like 14), 在4个组织中表达量较低(1.8~4.3)。(S-腺苷-L-甲硫氨酸依赖的甲基转移酶超家族蛋白)、(有机阳离子/肉碱转运体7)、(苏氨酸合酶1)、和在4个组织中不表达或表达量较低。
3 讨论
全基因组关联分析是以连锁不平衡为基础来挖掘覆盖全基因组遗传变异的一种方法。目前, GWAS已发展了大量统计方法, 例如MLM、FarmCPU、CMLM、MLMM和Fast-LMM (factored spectrally transformed linear mixed models)等[16-17]。本研究利用FarmCPU、CMLM和MLMM方法对穗轴粗进行关联分析, 发现FarmCPU的拟合效果最好, CMLM和MLMM方法拟合效果不理想(图2、附图1和附图2)。Liu等[18]利用FarmCPU、MLM和CMLM方法对玉米籽粒性状进行分析时, 也发现FarmCPU方法的检测功效最高。
本研究共发现12个穗轴粗显著关联SNP (<8.6E-07)。其中, 9个SNP位于控制产量、穗部相关性状(穗粒数、穗行数、行粒数、穗长、穗粗、轴重和轴粗)和籽粒相关性状(粒长、粒宽、粒厚和百粒重)的QTL区间内。S1_29006330在Zhang等[10]鉴定的1个穗轴粗QTL (qCD1-1)区间内。S1_29006330解释穗轴粗的变异率为13.10%, 为主效SNP。因此, S1_29006330可能是影响穗轴粗的重要位点, 可用于分子标记辅助育种。S4_83821463是控制穗轴粗的主效SNP (PVE=12.83%), 位于bin4.05, 该区域内检测到1个控制穗长的QTL[1], 1个控制行粒数的QTL[1], 1个控制穗行数的QTL[10]和1个控制产量、穗部性状和籽粒大小性状的元QTL (MetaQTL-27)[15]。S2_170889116也是控制穗轴粗的主效SNP, 其PVE最高, 为14.17%, 位于bin2.06。该区域Choe等[1]检测到1个同时控制单穗重和百粒重的QTL。主效SNP S2_204602646 (PVE=10.23%)在单穗粒重QTL qKWPE2-5区间内(201,971,890~209,300,839)[8]和控制穗部性状和籽粒大小性状的MetaQTL-13内[18]。主效SNP S4_83821463、S2_170889116和S2_204602646均在整合穗轴粗的元QTL内, 说明这3个主效SNP也可能是影响穗轴粗的重要位点。S3_170875493的表型变异解释率较高, 为8.55%, 位于bin3.06区域内。Upadyayula等[5]在该区域内鉴定到1个控制行粒数QTL。此外, 微效位点S5_17720377位于bin5.03, 该区域内Choe等[1]检测到1个同时控制穗轴重和百粒重的QTL。Yi等[8]在bin5.03区域内检测1个穗行数QTL qRN5。S5_17720377还位于控制穗部相关性状和籽粒性状的MetaQTL-32[19]内。S2_170090146位于Chen等[19]通过元分析整合的MetaQTL-11区间内, 该区间影响产量、穗部和籽粒大小相关性状。在S1_143964620和S4_29277313相关的bin区段内, Choe等[1]分别检测到1个粒长QTL和1个穗粒数QTL。不管是主效位点还是微效位点, 穗轴粗显著关联位点主要与已定位的穗部性状QTL区间存在重叠, 这说明穗轴粗可能是穗部多个性状共同调控的产物。
12个穗轴粗显著关联位点共检测到17个候选基因。编码细胞壁连接类受体, 为主效SNP S1_29006330的候选基因。WAK是植物类受体激酶, 由胞外域、跨膜域和胞内域组成, 能够跨越质膜, 使细胞识别并对外部环境进行响应[20]。转录本沉默会导致植株矮化、根发育受损和花粉败育[21]。WAKL具有典型的WAK结构, 可能参与花粉发育等穗发育相关的过程。根据maizeGDB的转录表达数据, 尽管在16~19 d的分生组织和雌小穗表达量较低(3.8~4.3), 但高于其余20个组织(附图3)。而且, 该基因对应的SNP S1_29006330在已定位的穗轴粗QTL区间内[10]。这表明,()可能是穗轴粗的候选基因。
编码转录因子, 为主效SNP S4_83821463挖掘的候选基因。ZIM具有GATA类锌指结构域即C-X2-C-X20-C-X2-C, 是一类植物特异的GATA转录因子[22]。Zhang等[10]利用连锁分析和全基因组关联分析挖掘的1个穗粗候选基因是转录因子。在16~19 d的分生组织和花丝中表达量较高(17.0~17.1) (附图3)。因此,()可能是影响穗轴粗的重要基因。S4_83821463挖掘的另一个基因, 编码高速泳动族蛋白-Y。其在16~19 d的分生组织、2~4 mm穗原基、6~8 mm穗原基和雌小穗中表达量较高(61.5~107.5) (附图3)。与SQUAMOSA启动子绑定蛋白类家族(SQUAMOSA promoter-binding protein-LIKE, SPL)结合形成异二聚体, 参与调节楸树的花器官发育, 但并不参与调节花期[23]。玉米SBP转录因子unbranched 2 ()和是SQUAMOSA启动子绑定蛋白家族成员, 通过调节侧原基和花序发育, 影响穗行数和雄穗分支数[24-25]。因此,()也可能是影响穗轴粗的重要基因。
S2_170889116是解释穗轴粗变异最高的1个主效SNP, 其挖掘的候选基因为(核孔复合蛋白)。该基因在16~19 d的分生组织、2~4 mm穗原基和6~8 mm穗原基的表达量较高(38.0~61.7)。核孔复合蛋白是核膜上的运输通道, 在染色质组装和基因表达中发挥重要作用[26]。但目前, 尚没有该类蛋白参与穗发育相关过程的研究报道。主效SNP S2_204602646挖掘的候选基因(组蛋白-赖氨素N-甲基转移酶)在16~19 d的分生组织、2~4 mm穗原基和6~8 mm穗原基的表达量均较高(97.0~119.2) (表2和附图3)。突变体表现出矮化和结实率低[27]。而且, 从花发育的十二时期开始, 相比正常植株, 该突变体的雄蕊发育迟缓[27]。因此,()可能是影响穗轴粗的重要候选基因。
此外, Wang等[28]通过整合产量相关性状全基因组关联分析和元分析结果, 发现1个候选基因编码木葡聚糖内糖基转移酶。木葡聚糖内糖基转移酶/水解酶是一类细胞壁松弛因子, 通过调节细胞壁弹性和延伸性来影响植物的生长发育和响应逆境胁迫等[29]。木葡聚糖内糖基转移酶/水解酶蛋白在过表达植株中表达上调, 从而导致了下胚轴和叶柄的延伸[30]。因此,()与一样可能是控制穗轴粗的重要基因。穗轴粗候选基因的挖掘为下一步精细定位和克隆提供信息, 为解析玉米穗轴粗遗传机理提供研究基础。
4 结论
发现12个与穗轴粗显著关联的SNP (<8.60E-07)。其中, 主效SNP 4个, 解释穗轴粗的变异率为10.23%~14.17%。挖掘候选基因17个, 其中、转录因子、、组蛋白-赖氨素N-甲基转移酶和可能是影响穗轴粗的重要基因。
[1] Choe E, Torbert R R. Genetic and QTL analysis of pericarp thickness and ear architecture traits of Korean waxy corn germplasm., 2012, 183: 243–260.
[2] Guo J, Chen Z, Liu Z, Wang B, Song W, Li W, Chen J, Dai J, Lai J. Identification of genetic factors affecting plant density response through QTL mapping of yield component traits in maize (L.)., 2011, 182: 409–422.
[3] Su C, Wang W, Gong S, Zuo J, Li S, Xu S. High density linkage map construction and mapping of yield trait QTLs in maize () using the genotyping-by-sequencing (GBS) technology., 2017, 8: 706–719.
[4] Zhu X M, Shao X Y, Pei Y H, Guo X M, Li J, Song X Y, Zhao M A. Genetic diversity and genome-wide association study of major ear quantitative traits using high-density SNPs in maize., 2018, 9: 966–981.
[5] Upadyayula N, da Silva H S, Bohn M O, Rocheford T R. Genetic and QTL analysis of maize tassel and ear inflorescence architecture., 2006, 112: 592–606.
[6] Zhao Y, Su C. Mapping quantitative trait loci for yield-related traits and predicting candidate genes for grain weight in maize., 2019, 9: 16112–16121.
[7] Jansen C, Lübberstedt T. Turning maize cobs into a valuable feedstock., 2012, 5: 20–31.
[8] Yi Q, Liu Y, Hou X, Zhang X, Li H, Zhang J, Liu H, Hu Y, Yu G, Li Y, Wang Y, Huang Y. Genetic dissection of yield-related traits and mid-parent heterosis for those traits in maize (L.)., 2019 19: 392–411.
[9] 王帮太, 吴建宇, 丁俊强, 席章营. 玉米产量及产量相关性状QTL的图谱整合. 作物学报, 2009, 35: 1836−1843. Wang B T, Wu J Y, Ding J Q, Xi Z Y. Map integration of QTLs for grain yield and its related traits in maize., 2009, 35: 1836−1843.
[10] Zhang X, Guan Z, Li Z, Liu P, Ma L, Zhang Y, Pan L, He S, Zhang Y, Li P, Ge F, Zou C, He Y, Gao S, Pan G, Shen Y. A combination of linkage mapping and GWAS brings new elements on the genetic basis of yield-related traits in maize across multiple environments., 2020, 33: 2881−2895.
[11] Meng L, Li H H, Zhang L Y, Wang J K. QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations., 2015, 3: 269–283.
[12] Liu X, Huang M, Fan B, Buckler E S, Zhang Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies., 2016, 12: e1005767.
[13] Segura V, Vilhjálmsson B J, Platt A, Korte A, Seren Ü, Long Q, Nordborg M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations., 2012, 44: 825−830.
[14] Zhang Z, Ersoz E, Lai C Q, Todhunter R J, Tiwari H K, Gore M A, Bradbury P J, Yu J, Arnett D K, Ordovas J M, Buckler E S. Mixed linear model approach adapted for genome-wide association studies., 2010, 42: 355−360.
[15] Pandis N. Linear regression., 2016, 149: 431−434.
[16] Lipka A E, Tian F, Wang Q, Peiffer J, Li M, Bradbury P J, Gore M A, Buckler E S, Zhang Z. GAPIT: genome association and prediction integrated tool., 2012, 28: 2397–2399.
[17] Lippert C, Listgarten J, Liu Y, Kadie C M, Davidson R I, Heckerman D. FaST linear mixed models for genome-wide association studies., 2011, 8: 833–835.
[18] Liu M, Tan X, Yang Y, Liu P, Zhang X, Zhang Y, Wang L, Hu Y, Ma L, Li Z, Zhang Y, Zou C, Lin H, Gao S, Lee M, Lubberstedt T, Pan G, Shen Y. Analysis of the genetic architecture of maize kernel size traits by combined linkage and association mapping., 2020, 18: 207–221.
[19] Chen L, An Y, Li Y X, Li C, Shi Y, Song Y, Zhang D, Wang T, Li Y. Candidate loci for yield-related traits in maize revealed by a combination of metaQTL analysis and regional association mapping., 2017, 22: 2190–2202.
[20] Kanneganti V, Gupta A K. Wall associated kinase from plants—an overview., 2008, 14: 109–118.
[21] Kanneganti V, Gupta A K. RNAi mediated silencing of a wall associated kinase, OsiWAK1 inresults in impaired root development and sterility due to anther indehiscence: wall associated kinases from., 2011, 17: 65–77.
[22] Shikata M, Takemura M, Yokota A, Kohchi T.ZIM, a plant-specific GATA factor, can function as a transcriptional activator., 2003, 67: 2495–2497.
[23] Wang Z, Zhu T, Ma W, Fan E, Lu N, Ou-Yang F, Wang N, Yang G, Kong L, Qu G, Zhang S, Wang J. Potential function of CbuSPL and gene encoding its interacting protein during flowering in., 2020, 20: 105.
[24] Chuck G S, Brown P J, Meeley R, Hake S. Maize SBP-box transcription factors unbranched2 and unbranched3 affect yield traits by regulating the rate of lateral primordia initiation., 2014, 111: 18775–18780.
[25] Du Y, Liu L, Peng Y, Li M, Li Y, Liu D, Li X, Zhang Z.expression and inflorescence development is mediated byand the distal enhancer, KRN4, in maize., 2020, 16: e1008764.
[26] Doucet C M, Hetzer M W. Nuclear pore biogenesis into an intact nuclear envelope., 2010, 119: 469−477.
[27] Chen L Q, Luo J H, Cui Z H, Xue M, Wang L, Zhang X Y, Pawlowski W P, He Y. ATX3, ATX4, and ATX5 encode putative H3K4 methyltransferases and are critical for plant development., 2017, 174: 1795–1806.
[28] Wang Y, Wang Y, Wang X, Deng D. Integrated meta-QTL and genome-wide association study analyses reveal candidate genes for maize yield., 2020, 39: 229–238.
[29] Miedes E, Suslov D, Vandenbussche F, Kenobi K, Ivakov A, Van Der Straeten D, Lorences E P, Mellerowicz E J, Verbelen J P, Vissenberg K. Xyloglucan endotransglucosylase/hydrolase (XTH) overexpression affects growth and cell wall mechanics in etiolatedhypocotyls., 2013, 64: 2481–2497.
[30] Shikata M, Matsuda Y, Ando K, Nishii A, Takemura M, Yokota A, Kohchi T. Characterization ofZIM, a member of a novel plant-specific GATA factor gene family., 2004, 55: 631–639.
附图1 CMLM方法不同环境穗轴粗(CD)全基因组关联分析的曼哈顿图和QQ图
Fig. S1 Manhattan plots and quantile-quantile plots for genome-wide association analysis for ear cob diameter using CMLM method in different environments
DC、YC、SY、YY分别表示郸城、虞城、三亚和原阳。2017和2019代表年份。
DC, YC, SY, and YY represent Dancheng, Yucheng, Sanya, and Yuanyang, respectively. 2017 and 2019 denote in 2017 and in 2019.
附图2 MLMM和FarmCPU方法没有检测到显著穗轴粗SNP的曼哈顿图和QQ图
Fig. S2 Manhattan plots and quantile-quantile plots for no significant SNP for ear cob diameter using MLMM and FarmCPU
DC、YC、SY、YY分别表示郸城、虞城、三亚和原阳。2017和2019代表年份。
DC, YC, SY, and YY represent Dancheng, Yucheng, Sanya, and Yuanyang, respectively. 2017 and 2019 denote in 2017 and in 2019.
附图3 maizeGDB中17个候选基因在不同组织的表达情况
Fig. S3 Expression profiles of 17 candidate genes in different tissues retrieved from maize GDB
Genome-wide association study of ear cob diameter in maize
MA Juan, CAO Yan-Yong, and LI Hui-Yong*
Institute of Cereal Crops, Henan Academy of Agricultural Sciences, Zhengzhou 450002, Henan, China
Maize ear cob diameter is an important trait impacting the yield of grain and cob, and the analysis of its genetic mechanism will provide a guidance for high-yield breeding. In this study, the genotypes of 309 inbred lines were identified by genotyping-by-sequencing technology. FarmCPU (fixed and random model circulating probability unification), MLMM (multiple loci mixed linear model), and CMLM (compressed mixed linear model) were used to identify significant single nucleotide polymorphisms (SNP) for ear cob diameter of Yuanyang of Henan province, Dancheng of Henan province, Yucheng of Henan province, Sanya of Hainan province in 2017 and 2019, and best linear unbiased estimate environment. A total of 12 significant SNP for ear cob diameter were detected at< 8.60E-07. S4_29277313 was detected from Yuanyang in 2017 using FarmCPU and MLMM. The phenotypic variance explained of S1_29006330, S2_170889116, S2_2046026464, and S4_83821463 ranged from 10.23% to 14.17%, and were considered major-effect SNP. In addition, S1_29006330 was mapped in the interval of known QTL for ear cob diameter. A total of 17 candidate genes were identified. Among them,(wall-associated receptor kinase-like 14), transcription factor(zinc-finger protein expressed in inflorescence meristem 35),(HMG-Y-related protein A), histone-lysine N-methyltransferase(trithorax 4), and(xyloglucan endotransglucosylase/hydrolase protein 32) might be important genes for ear cob diameter. The identification of four major-effect SNP and five candidate genes can provide an information for molecular marker-assisted breeding, fine mapping, and gene cloning.
maize; genome-wide association study (GWAS); FarmCPU; ear cob diameter
10.3724/SP.J.1006.2021.03048
本研究由河南省科技攻关项目(182102110368)和河南省农业科学院优秀青年基金(2020YQ04)资助。
This study was supported by the Science and Technology Project of Henan Province (182102110368) and the Science-Technology Foundation for Outstanding Young Scientists of Henan Academy of Agricultural Sciences (2020YQ04).
李会勇, E-mail: lihuiyong1977@126.com
E-mail: majuanjuan85@126.com
2020-08-14;
2020-12-01;
2021-01-04.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20210104.1147.004.html
