反相高效液相色谱测定枸橼酸托法替布有关物质的方法学研究
2021-02-26郭颖昕陈伟燕石海芹郭燕燕
张 婷 郭颖昕 陈伟燕 石海芹 郭燕燕
上海上药中西制药有限公司 (上海 201806)
枸橼酸托法替布又名枸橼酸托法替尼,即(3R,4R)-4-甲基-3-(甲基-7H-吡咯并[2,3-d]嘧啶-4-基氨基)-β-氧代-1-哌啶丙腈枸橼酸盐(C16H20N6O·C6H8O7),其化学结构如图1 所示。
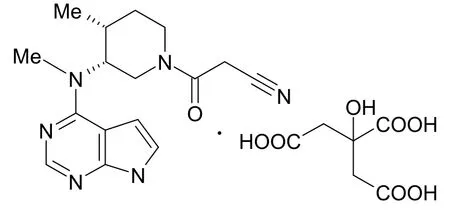
图1 枸橼酸托法替布的化学结构
类风湿性关节炎(RA)是一种由关节滑膜慢性炎症、抗瓜氨酸肽抗体和类风湿因子的异常表达介导的慢性系统免疫性疾病,其临床症状主要为滑膜增生、血管翳的形成、软骨损失和关节破坏等,如不及时给予规范治疗,病情可逐渐加重,最后导致畸形、关节强直和功能丧失,是临床常见的主要致残性疾病之一。我国类风湿性关节炎患病人数近500 万;该病致残率高,给社会和患者均造成沉重的负担。
目前,针对类风湿性关节炎的治疗有很多种,主要包括非甾体抗炎药、糖皮质激素类药物、慢作用抗风湿药、生物药物及小分子激酶(JAK)抑制剂。JAK 属于胞内酪氨酸激酶家族,能够从细胞因子、干扰素和多种激素受体向细胞核传递信号,从而合成多种生物活性物质,影响细胞的代谢和功能,因此,激酶功能障碍则会导致造血功能障碍和免疫功能低下。在类风湿性关节炎患者的治疗过程中,选用JAK抑制剂可以迅速有效地抑制炎症细胞因子的过度表达,从而达到治疗目的[1]。
枸橼酸托法替布是第一个被批准用于人类自身免疫性疾病的小分子非受体型酪氨酸蛋白激酶抑制剂。枸橼酸托法替布片于2012 年在美国首先批准上市,目前已在日本、俄罗斯、澳大利亚、加拿大等全球50 多个国家和地区获得上市批准。2017 年,欧洲药品管理局(EMA)也批准该药用于类风湿性关节炎的治疗,并对近5 000 名患者进行随访,其长期安全性和有效性数据得到了进一步证实。枸橼酸托法替布的临床疗效显著,且可与甲氨蝶呤或其他非生物改善病情抗风湿药(DMARD)联合使用,不良反应轻微、安全性高、服用方便,深受医生和患者的青睐[2-3],是近年世界范围内销售量增长较快的药物之一。
枸橼酸托法替布为化学药物3 类药。目前,对于枸橼酸托法替布有关物质的测定还不够全面。根据其合成工艺及降解机制,选取了9 种主要的已知杂质 ,包括工艺杂质 TZ1、TZ3、TZ4、TZ5、PF-05211077、PF-05287430 以及降解杂质TZ2、PF-05579970、P-05091895。为了进一步控制上海上药中西制药有限公司产品枸橼酸托法替布片的质量,参考国内外相关文献[1-7],系统地研究分析了枸橼酸托法替布有关物质9 种已知杂质的测定,并采用C18色谱柱、利用反相高效液相色谱(RP-HPLC)建立了枸橼酸托法替布有关物质的测定方法。依据外标法测定4 种已知杂质的含量,采用不加校正因子的主成分自身对照法测定另外5 种已知杂质。
实验结果证实,该方法具有良好的专属性、耐用性、稳定性,以及较高的灵敏度[4-5],也进一步说明其适用于上海上药中西制药有限公司产品有关物质项的检测。
1 实验部分
1.1 仪器
AL104 电子天平、XPE105 电子天平,梅特勒-托利多国际贸易有限公司;1260 全自动高效液相色谱仪,安捷伦科技有限公司。
1.2 试剂
三氟乙酸,国药集团化学试剂有限公司;甲醇、乙腈,默克股份两合公司;癸烷磺酸钠,赛默飞世尔科技(中国)有限公司。所用试剂均为色谱纯。
1.3 标准品
实验过程中所使用的标准品信息如表1 所示。
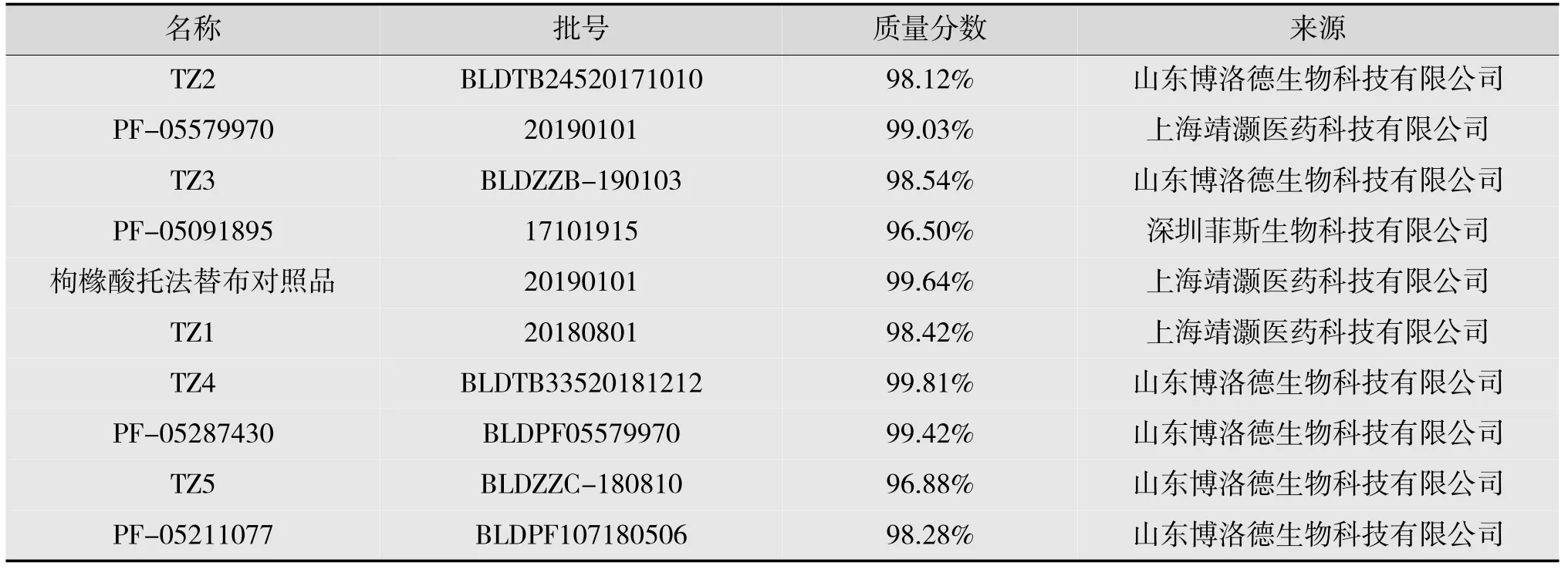
表1 标准品信息
1.4 方法
1.4.1 色谱条件
用十八烷基硅烷键合硅胶为填充剂[CAPCELL PAK C18MG(4.6 mm×150 mm,3 μm)或效能相当的色谱柱],加装捕集柱(Ghost-Buster,4.6×50 mm);以磷酸盐缓冲液(取磷酸二氢钾1.36 g,加水溶解并稀释至1 000 mL,加入1 mL 0.5 mg/mL 的癸烷磺酸钠溶液,混匀)为流动相A,乙腈为流动相B,梯度洗脱。梯度程序如表2 所示。洗脱流速为0.8 mL/min,柱温箱温度为45 ℃,紫外检测波长为216 nm。
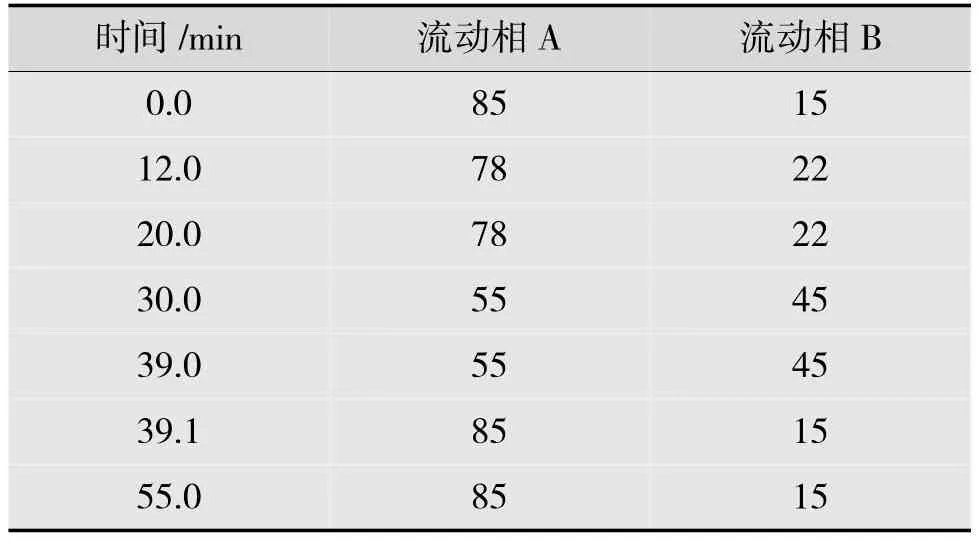
表2 梯度洗脱比例
1.4.2 溶液的配制
(1)系统适用性溶液的制备
取枸橼酸托法替布对照品及各杂质对照品适量,配制成每1 mL 中含托法替布0.15 mg,TZ1、TZ2、TZ3、TZ4、TZ5 各0.8 μg,PF-05579970、PF-05091895、PF-05287430、PF-05211077 各0.375 μg的混合溶液作为系统适用性溶液。
(2)供试品溶液的制备
称取本品研磨细粉适量(约相当于托法替布15 mg),置于100 mL 容量瓶中;加乙腈-0.05%三氟乙酸(二者体积比为5∶95,下同)溶液振摇使托法替布溶解,放冷;加乙腈-0.05%三氟乙酸溶液稀释至刻度,摇匀,滤过,作为供试品溶液。
(3)对照溶液的制备
精密量取供试品溶液适量,以乙腈-0.05%三氟乙酸溶液定量稀释,制成每1 mL 中含0.75 μg 托法替布的溶液作为对照溶液。
(4)灵敏度溶液的制备
精密量取供试品溶液适量,以乙腈-0.05%三氟乙酸溶液定量稀释,制成每1 mL 中含0.03 μg 托法替布的溶液作为灵敏度溶液。
(5)混合对照品溶液的制备
取杂质TZ3、TZ4、PF-05091895、PF-05211077对照品适量,精密称定,用乙腈-0.05%三氟乙酸溶液定量稀释,制成每1 mL中含杂质TZ3、TZ4、PF-05091895、PF-05211077 各0.3 μg 的混合溶液作为混合对照品溶液。
1.4.3 测定方法
分别取系统适用性溶液及灵敏度溶液各20 μL注入液相色谱仪,考察分离度并调节检测灵敏度使主峰峰高的信噪比大于10;分别取供试品溶液与对照品溶液各20 μL 注入液相色谱仪,记录色谱图至主峰保留时间的3 倍。采用外标法以峰面积计算供试品杂质TZ3、TZ4、PF-05091895、PF-05211077 的含量,采用自身对照法计算供试品杂质PF-05579970、PF-05287430 、TZ1、TZ2、TZ5 的含量。
2 结果与讨论
2.1 系统适用性试验
取系统适用性溶液20 μL,注入液相色谱仪,记录色谱图,结果如表3 所示。在有关物质色谱条件下,理论板数按托法替布计为20 323,各峰间的分离度均大于2.1,说明该法色谱条件分离效果良好。
2.2 检测限(LOD)、定量限(LOQ)试验
精密称取枸橼酸托法替布标准品及各杂质适量,以乙腈-0.05%三氟乙酸溶液逐级稀释,注入液相色谱仪,调节检测灵敏度以主成分峰的峰高与基线噪音的信噪比大于或等于10 时对应的质量浓度为定量限;将定量限质量浓度继续稀释,注入液相色谱仪,调节检测灵敏度,以主成分峰峰高与基线噪音的信噪比大于或等于3 时对应的质量浓度为检测限。由表4 结果可见,在该色谱条件下,各杂质检测限均在供试品质量浓度1‰之下,说明该色谱法灵敏度较高。
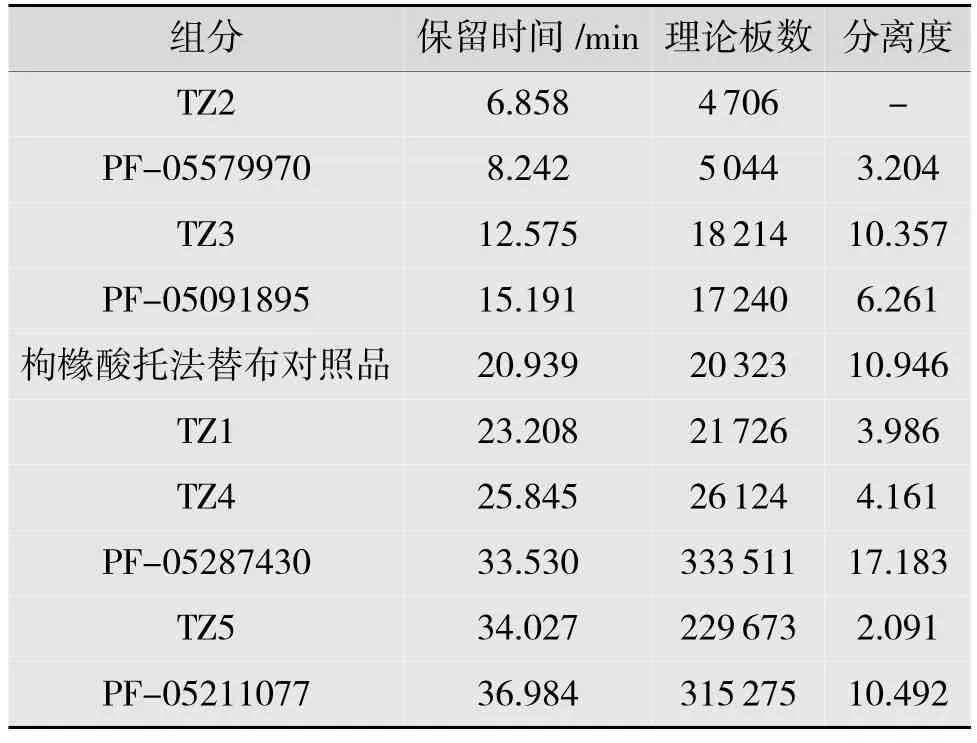
表3 有关物质-系统适用性试验结果
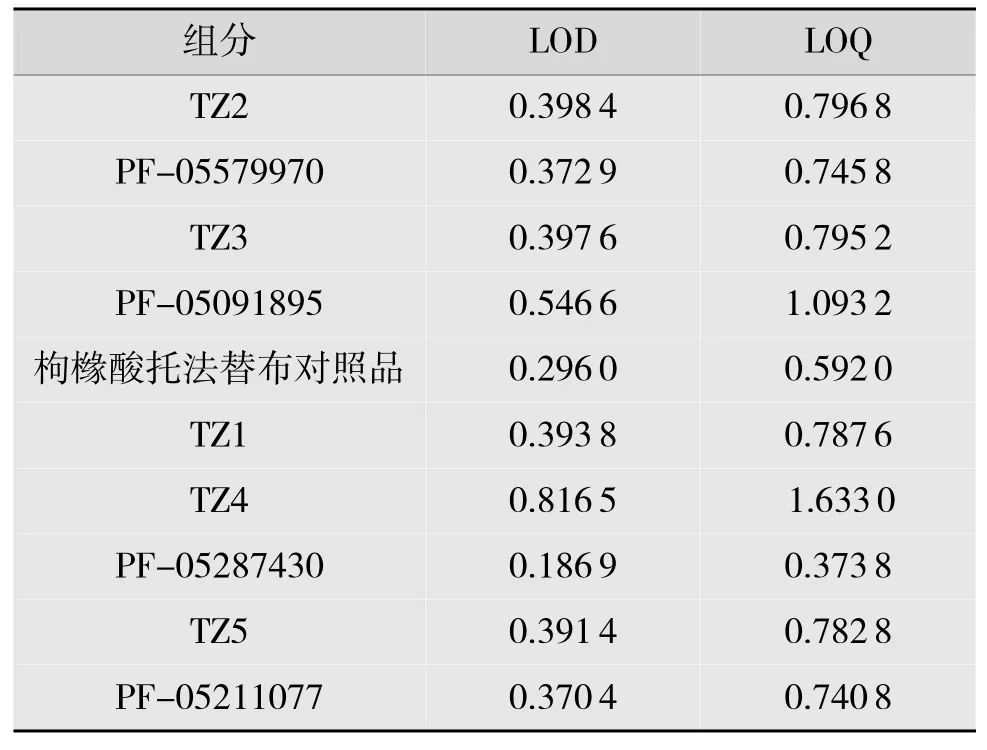
表4 有关物质的检测限与定量限ng/mL
2.3 专属性考察
称取空白辅料约600 mg,置于100 mL 容量瓶中,加入乙腈-0.05%三氟乙酸溶液适量,超声处理至分散均匀,放冷,定容、摇匀、滤过,取续滤液作为空白辅料溶液;称取空白包衣材料约18 mg,置于100 mL 容量瓶中,加入乙腈-0.05%三氟乙酸溶液适量,超声处理至分散均匀,放冷,定容、摇匀、滤过,取续滤液作为空白包衣溶液;分别精密量取20 μL空白辅料溶液、空白包衣溶液注入液相色谱仪,结果表明辅料及包衣材料均不干扰样品的检测。
2.4 线性试验
精密称取枸橼酸托法替布及各杂质对照品适量,分别用乙腈-0.05%三氟乙酸溶液稀释制成一系列不同质量浓度溶液,在有关物质色谱条件下,分别精密量取20 μL,注入液相色谱仪,记录色谱图,按上述同法进行线性回归,结果如表5 所示。
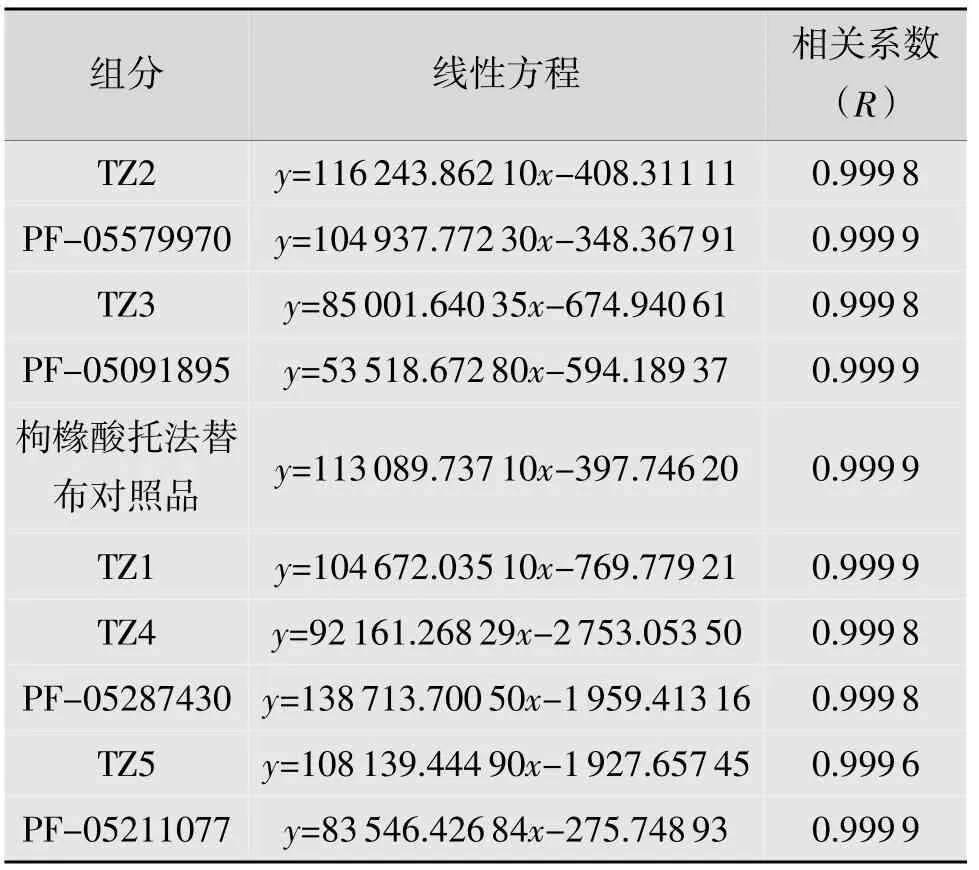
表5 枸橼酸托法替布及各杂质线性试验结果
根据结果,TZ2 在0.039 8~1.593 5 μg/mL(相当于供试品质量浓度的0.027%~1.000%)范围内,线性关系良好;PF-05579970 在0.037 3~1.491 6 μg/mL(相当于供试品质量浓度的0.025%~1.000%)范围内,线性关系良好;TZ3 在0.039 8~1.590 4 μg/mL(相当于供试品质量浓度的0.027%~1.000%)范围内,线性关系良好;PF-05091895 在0.054 7~1.457 5 μg/mL(相当于供试品质量浓度的0.036%~1.000%)范围内,线性关系良好;托法替布在0.029 6~1.480 1 μg/mL(相当于供试品质量浓度的0.020%~1.000%)范围内,线性关系良好;TZ1 在0.039 4~1.575 1 μg/mL(相当于供试品质量浓度的0.027%~1.000%)范围内,线性关系良好;TZ4 在0.081 7~1.633 1 μg/mL(相当于供试品质量浓度的0.054%~1.000%)范围内,线性关系良好;PF-05287430 在0.018 7~1.494 9 μg/mL(相当于供试品质量浓度的0.012%~1.000%)范围内,线性关系良好;TZ5 在0.039 1~1.565 6 μg/mL(相当于供试品质量浓度的0.026%~1.000%)范围内,线性关系良好;PF-05211077 在0.037 0~1.481 5 μg/mL(相当于供试品质量浓度的0.025%~1.000%)范围内,线性关系良好。
2.5 溶液稳定性试验
取本品内容物适量,按有关物质测定法制备供试品溶液,在室温(不避光)下放置48 h 后,精密量取20 μL 注入液相色谱仪,记录色谱图,考察供试品溶液的稳定性。结果表明,供试品溶液室温下放置48 h 内,各杂质含量(质量分数,下同)与0 h 含量的绝对差值为0~0.02%,总杂含量的绝对差值为0.02%。因此,供试品溶液在室温下48 h 内稳定。
2.6 重复性试验
取本品适量,按上述方法制备供试品溶液及对照品溶液。同法操作6 份,按上述方法检查有关物质,计算重复性试验结果。6 份供试品溶液中:TZ2含量的均值为0.04%,相对标准偏差(RSD)为0;平均相对保留时间(RRT)为0.68 的杂质含量的均值为0.01%,RSD 为0;RRT 为0.77 的杂质含量的均值为0.01%,RSD 为0;RRT 为1.69 的杂质含量的均值为0.02%,RSD 为0.01%;RRT 为1.90 的杂质含量的均值为0.02%,RSD 为0;RRT 为2.13 的杂质含量的均值为0.02%,RSD 为0;PF-05091895 含量的均值为0.03%。6 份供试品溶液的绝对差值为0.01%,总杂含量的RSD 为4.23%,表明该测定方法具有优异的重复性。
2.7 准确度试验
分别精密称取各杂质10 mg 置于1 000 mL 容量瓶中,各加乙腈-0.05%三氟乙酸溶液溶解并稀释定容至刻度,摇匀,作为各个杂质储备液。按处方量分别加入枸橼酸托法替布及空白辅料;每种杂质分别设计3 个不同的质量浓度,各制成每1 mL 分别含杂质约0.1,0.4 和0.5 ng 的混合溶液。每个质量浓度分别配制3 份供试品溶液,按上述方法进行回收率测定,结果如表6 所示。采用该法测定的枸橼酸托法替布各杂质9 份样品平均回收率均在97%~104%之间,RSD 均符合接受标准,表明该方法准确度高,也进一步证实了该测定方法的精确性与可靠性良好。
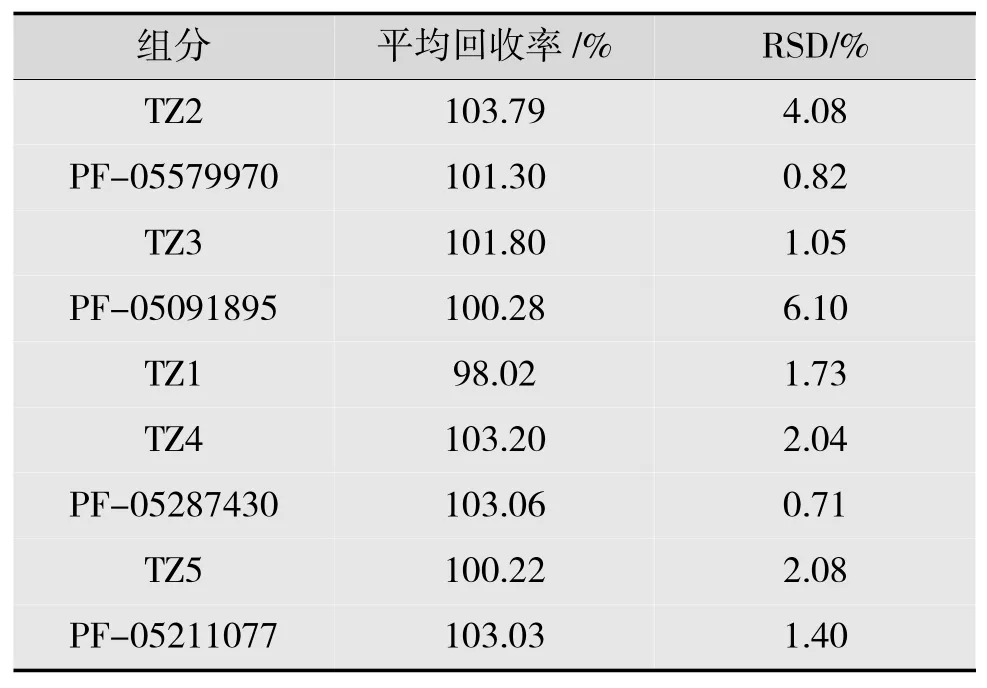
表6 有关物质回收率试验结果
2.8 耐用性试验
按前文所述配制系统适用性溶液,按照相关测定方法,每次只改变一个色谱条件,考察该方法的耐用性,结果如表7 所示。
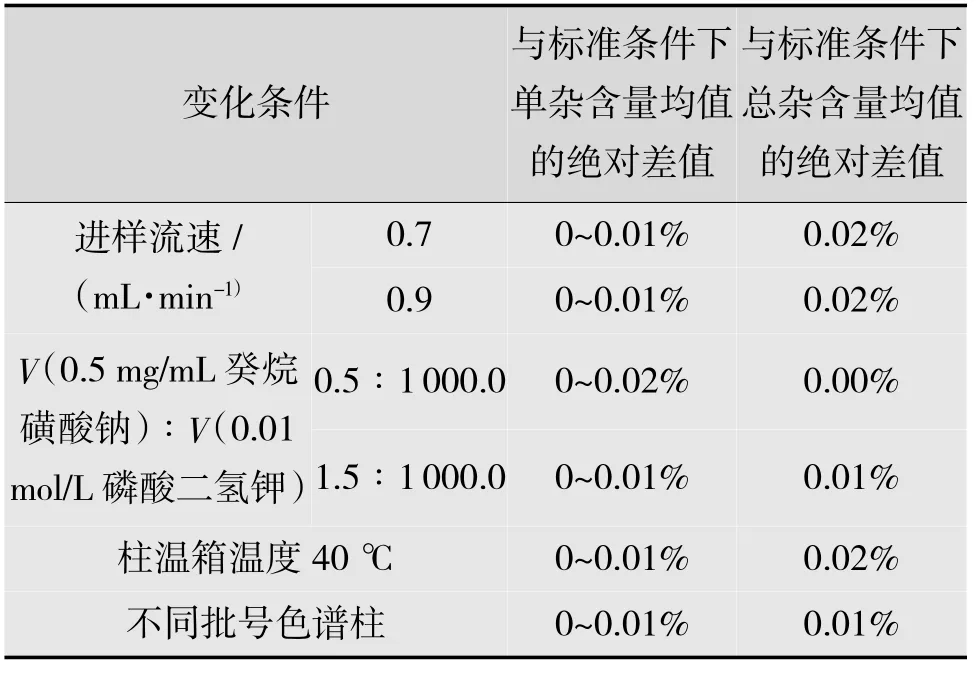
表7 有关物质耐用性试验结果
由表7 可知,进样流速、离子对试剂比例、色谱柱批号、柱温箱温度等在一定程度下均不影响主药的测定,表明该方法的系统耐用性良好。
3 结语
以十八烷基硅烷键合硅胶为填充剂[CAPCELL PAK C18MG(4.6 mm×150 mm,3 μm)或效能相当的色谱柱],以0.01 mol/L 磷酸盐缓冲液-乙腈为流动相梯度洗脱,建立反相高效液相色谱法对枸橼酸托法替布片有关物质进行检测,该法灵敏度高、杂质分离效果好、耐用性优异,溶液质量浓度与峰面积相关性显著,处方中辅料不干扰药物含量的测定,且操作简便、检测迅速、准确度高,适用于枸橼酸托法替布有关物质的检测。
