一种利用反相高效液相色谱法测定乳酸菌发酵液中苯乳酸的方法及其评价
2020-12-10林毅侃谢佳雨李柏林
张 雯,林毅侃,谢佳雨,欧 杰,3,4,*,李柏林
(1.上海海洋大学食品学院,上海 201306;2.上海市质量监督检验技术研究院/国家食品质量监督检验中心(上海),上海 200233;3.上海水产品加工及贮藏工程技术研究中心,上海 201306;4.农业部水产品贮藏保鲜质量安全风险评估实验室,上海 201306)
苯乳酸(Phenyllactic Acid,PLA)是一种小分子化合物,抑菌谱广,溶解性好,易在食物系统中扩散,在较宽pH范围内能够保持稳定[1]。苯乳酸能够存在于新西兰麦卢卡蜂蜜[2-3]、泡菜[4]和发酵面团[5],对人体无毒无害。因此,苯乳酸有望开发成为一种新型食品抑菌剂。
苯乳酸能抑制某些革兰氏阴性菌[6]、革兰氏阳性菌[7-8]、真菌[9]和真菌毒素[10]。苯乳酸能够通过影响微生物细胞膜从而抑制微生物的生长繁殖[11-12],有研究表明在超高温瞬时灭菌乳中添加1 mg/mL的苯乳酸可以显著抑制单增李斯特菌的生长[2]。乳酸菌由于一般公认安全(Generally Recognized as Safe,GRAS),其合成苯乳酸的能力已被广泛研究[13-14]。有研究在中式泡菜[4]和韩国泡菜[15]中分离到能够合成苯乳酸的植物乳杆菌(Lactobacillusplantarum)。有研究报道利用重组大肠杆菌合成苯乳酸[16],但乳酸菌合成苯乳酸更具有安全性,更能节省食品工业成本。
目前乳酸菌发酵液中苯乳酸的检测方法主要有HPLC法[17-18]、超高效液相色谱-串联质谱法[15](Ultra-High Performance Liquid Chromatography Mass Spectrometry,UPLC-MS/MS)、HPLC-MS/M法[3]、薄层层析法[19]和气相色谱法[20]。气相色谱法检测准确能够满足检测需要,但发酵液需冻干后酯化,前处理复杂,样品数量较大时,检测效率不高;UPLC-MS/MS法和HPLC-MS/MS法设备昂贵,成本较高,检测时间长;HPLC法由于前处理简单,准确度高,应用较为广泛。有研究对比了HPLC法检测乳酸菌发酵液中苯乳酸0.22 μm微滤后进样和微滤后过SPE柱对苯乳酸检测的影响,发现微滤后直接进样回收率更好[17]。
目前对可以用于HPLC法测定乳酸菌中的苯乳酸流动相和固定相各不相同,针对使用的流动相和固定相进行的讨论较少。选择合适的流动相和固定相可以提高检测效率,对检测方法进行方法学和不确定度评价,能够衡量检测结果的可信度。本实验旨在选择较为合适的固定相和流动相的组合,并对方法进行评价、计算不确定度,为乳酸菌合成苯乳酸的相关研究及检测提供参考。
1 材料与方法
1.1 材料与仪器
短乳杆菌P3(LactobacillusbrevisP3) 分离自泡菜,由实验室-80 ℃保存;植物乳杆菌ATCC014(L.plantarumATCC8014)中国普通微生物菌种保藏管理中心;苯乳酸标准品 纯度≥98%,上海子起生物科技有限公司;乙腈、甲醇、TFA 色谱级,美国Fisher公司;MRS液体培养基 美国BD公司。
Agilent-1260高效液相色谱仪 美国Agilent公司;恒温水浴锅 天津欧诺仪器仪表有限公司;超声波水浴锅 江苏盛蓝仪器制造有限公司;雷磁PHS-3E型pH计 上海仪电科学仪器股份有限公司。
1.2 实验方法
1.2.1 标准储备液制备 准确称取苯乳酸0.01 g定容于10 mL容量瓶中,得到浓度为100 mg/L标准储备液,4 ℃避光存放。
取适量储备液,用基质空白液稀释成浓度分别为0.2、0.4、0.8、1.0、2.0、5.0 mg/L的标准工作液进行HPLC分析。
1.2.2 发酵液样品前处理 将L.brevisP3和L.plantarumATCC8014以1%接种量分别接种于MRS液体培养基,35 ℃下分别培养120 h。分别取两种菌株的发酵液10000 r/min下离心2~5 min,取上清液0.22 μm微滤后进样。
1.2.3 高效液相色谱测定条件的优化
1.2.3.1 不同固定相对苯乳酸高效液相色谱的影响 本实验测试了三种固定相,分别是:I:AgilentZorbax SB-C18色谱柱(4.6 mm×250 mm,5 μm),无封端色谱柱;II:AgilentEclipse XDB-C18色谱柱(4.6 mm×250 mm,5 μm),双封端色谱柱;III:Agilent Zorbax SB-C8色谱柱(4.6 mm×250 mm,5 μm)。
流动相为0.05%TFA-乙腈,体积比为75∶25,流速1 mL/min,检测波长210 nm,进样量10 μL,柱温30 ℃。
1.2.3.2 不同流动相对苯乳酸高效液相色谱的影响 以0.05% TFA为水相,1.2.3.1中得出的较优色谱柱为固定相,考察乙腈和甲醇对苯乳酸HPLC检测的影响,平衡30 min后,进样3次。
以乙腈为有机相,考察0.05% TFA水溶液、0.10%甲酸水溶液、0.10%磷酸水溶液、0.02 mol/L乙酸铵溶液、0.05 mol/L乙酸铵溶液、0.02 mol/L KH2PO4溶液和0.02 mol/L K2HPO4溶液为水相对苯乳酸的HPLC检测的影响,测试不同的水相对苯乳酸的保留能力和对峰型的影响。每种流动相组合试验3种体积比:15∶85、20∶80和25∶75,每种流动相比例下,平衡30 min后,进样3次。
1.2.4 方法学评价
1.2.4.1 线性关系、检出限和定量限的测定 通过HPLC分析6种浓度(0.2、0.4、0.8、1.0、2.0和5.0 mg/L)的标准品溶液,绘制苯乳酸的校准曲线,使用Aglient1260离线软件计算线性回归,以标准品浓度为x轴,以峰面积为y轴,计算回归方程和系数。
检出限定义为信噪比(Signal-Noise Ratio,RSN)为3时对应的质量浓度,定量限定义为RSN为10时对应的质量浓度。在1.2.3得到的色谱条件下,仪器平衡1 h后,仪器的基线噪声为0.27 mAu。
1.2.4.2 精密度和准确性的测定 精密度试验取2.0 mg/L标准溶液,在1.2.3得到的色谱条件下,在1和30 d分别连续进样6次,测定并计算峰面积平均值和RSD。
重复性试验制备6份2.0 mg/L标准溶液,在1和30 d分别进样,测定并计算峰面积平均值和RSD。
1.2.4.3 回收率实验 取L.brevisP3发酵液适当稀释后10000 r/min离心2 min,取上清液经过适当稀释后得到供试品,0.22 μm微滤后进样,得到发酵液中苯乳酸含量。
向供试品中分别加入低、中、高3个不同水平的的标准品(即加入折合质量为0.2、0.4和1.0 mg/L的苯乳酸标准品),按照1.2.3得到的色谱条件进行HPLC分析考察方法的回收率。
1.2.4.4 稳定性的测定 分别于1、10、20和30 d测定2.0 mg/L标准溶液的峰面积,并计算平均值和RSD。
1.2.5 方法的不确定度 由于样品为直接测定,不确定度的来源有配制标准溶液引入的不确定度、标准曲线引入的不确定度、重复性引入的不确定度,加标回收引入的不确定度,根据以上不确定度来源计算扩展不确定度。
1.2.5.1 配制标准溶液引入的不确定度(ustandard) 配制标准溶液的不确定度主要来自标准品(ustock)和标准溶液的配制(upreparation)。仪器的膨胀系数为2.1×10-4,温度变化范围为±4 ℃,10 mL容量瓶检定证书偏差为10±0.02 mL,近似于矩阵分布,由定容体积引起的不确定度ur(V1)=0.00125。1 mL移液管检定证书偏差为1±0.01 mL,近似于矩阵分布,由移液引起的不确定度ur(V2)=0.00579[21-22]。
式(1)
式(2)
式(3)
式中,p%:标准品纯度。
1.2.5.2 标准曲线引入的不确定度(ucalibration) 标准曲线为y=ax+b,a为斜率,b为截距。
式(4)
式(5)
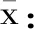
1.2.5.3 重复性引入的不确定度(urep)
式(6)
式中,Srep-重复性实验标准差,x0-重复性实验中所用标准溶液浓度,n-标准溶液测定重复数。
1.2.5.4 回收率引入的不确定度(urecovery)
式(7)

通过显著性确定平均回收率是否与1有显著性差异。检测统计数据t通过下式计算:
式(8)
t与95%置信度,n-1自由度的双边临界值tcrit比较(其中n是用来评估回收率平均值的测试结果的数目),假如t大于或等于tcrit值,则ˉrec与1有显著性差异,urecovery不计入合成不确定度,反之则计入合成不确定度,tcrit≈2.306。
1.2.5.5 相对合成不确定高度和扩展不确定度 根据不确定度的传播定律,相对合成不确定度为:
式(9)
当置信水平P=95%(k=2),相对扩展不确定度:
U=kc×urel
式(10)
1.3 数据处理
实验中每组平行重复6次,用Origin 2018作图,Agilent 1260离线软件和Microsoft Excel 365进行数据分析。
2 结果与分析
2.1 高效液相色谱条件的优化
2.1.1 不同固定相对苯乳酸高效液相色谱的影响 本实验采用RP-HPLC法,分别测试了三种色谱柱对苯乳酸的分离效果,分别是未封端C18色谱柱(Ⅰ)、封端C18色谱柱(Ⅱ)和C8色谱柱(Ⅲ)。色谱条件参照1.2.3.1。
色谱柱Ⅰ(图1a)和色谱柱Ⅱ(图1b)均能够在5.9 min平稳出峰,色谱柱I有轻微拖尾现象,色谱柱Ⅲ(图1c)拖尾现象严重,色谱柱Ⅱ相比色谱柱Ⅰ和Ⅲ峰型较好。根据色谱柱使用说明,C18和C8色谱柱适用pH范围为2.0~9.0。经测定,乳酸菌发酵液的最低pH为3.7,能够满足色谱柱的适用pH范围。色谱柱II出峰平稳,无拖尾,峰型对称,更适宜苯乳酸的HPLC检测。

图1 不同固定相对苯乳酸HPLC分析的影响
2.1.2 不同流动相对苯乳酸高效液相色谱的影响 流动相的优化以双封端C18色谱柱为固定相,首先以0.05% TFA为水相,考察了甲醇作为有机相在双封端的C18柱上对苯乳酸的洗脱能力。如图2a所示,以甲醇为有机相,在峰后出现了轻微拖尾现象。乙腈比甲醇具有更强的洗脱能力,以乙腈作为流动相峰型较好,有机相使用的比例小。因此,使用乙腈(图1b)作为有机相洗脱苯乳酸较好。

图2 不同溶剂系统对苯乳酸HPLC分析的影响
其次考察了不同水相对苯乳酸HPLC检测的影响。图2b表明,0.10%磷酸水溶液-乙腈为流动相,对苯乳酸的保留能力差。以0.02 mol/L乙酸铵溶液-乙腈为流动相时(图2c),峰前出现小峰,且峰形不佳。流动相为0.05 mol/L乙酸铵溶液-乙腈时,保留能力弱,出现了峰型分裂(图2d)。在以0.10%甲酸溶液-乙腈为流动相的条件下,在85∶15 (v/v)和80∶20 (v/v)(图2e,f),不同流动相比例下,峰形存在分裂和小峰的现象和前尾峰的现象,在75∶25 (v/v)下有轻微拖尾现象(图2g)。0.02 mol/L KH2PO4溶液-乙腈(图2h)和0.02 mol/L K2HPO4溶液-乙腈(图2i)峰型有拖尾现象。综上,0.05%TFA水溶液-乙腈为流动相时峰型更佳(图1b)。

表1 线性回归方程、检出限和定量限

表2 仪器精密度和重复性实验
2.2 方法学评价
2.2.1 线性范围、检出限及定量限 以2.1得出的色谱条件进行方法学评价,线性范围、检出限及定量限结果见表1,回归方程y=32.5x+1.04且r>0.990,该方法在0.2~5.0 mg/L内有良好的线性。使用0.2 mg/L的标准溶液测定检出限和定量限,分别为0.1400和0.4600 mg/L,表明该方法灵敏度高,能够满足检测需要。
2.2.2 仪器精密度和重复性实验 表2为仪器精密度和重复性,苯乳酸标准溶液在1 d精密度和重复性测试RSD分别为0.30%和0.64%,在30 d精密度和重复性测试RSD分别为0.59%和0.79%。结果表明,仪器精密度良好,方法具有良好的重现性。
2.2.3 回收率实验 表3为回收率实验结果,L.brevisP3发酵液稀释后中苯乳酸浓度为0.14 mg/L,加标回收率在99.88%~101.08%,在80%~110%之间[23],回收率较好,能够满足检测需要。

表3 回收率实验
2.2.4 稳定性实验 对30 d内该苯乳酸标准储备液稳定性进行了测试,如表4所示,在30 d内峰面积为69.73 mAu,且RSD为2.45%,4次稳定性测试结果差异不显著(P>0.05),峰面积测定结果稳定,结果表明,标准储备液在30 d内4 ℃保存下性质稳定。

表4 苯乳酸标准储备液稳定性
2.3 不确定度评价
实验操作中不可避免的产生不确定度,不确定度能够反映结果的可靠性,本实验计算了L.brevisP3发酵液中苯乳酸测定的扩展不确定度,其中包括标准溶液的配制,标准曲线、重复性和回收率。回收率不确定度为0.0014,t大于tcrit值,回收率不计入相对不确定度。
标准溶液配制的不确定度为0.013,标准曲线的不确定度为0.0098,重复性的不确定度为0.0015,扩展不确定度为0.0046。在不确定度来源中,标准溶液稀释和校准的不确定度是整体不确定度中重要的来源。重复性引入的不确定度最小,因此在配制标准溶液和建立标准曲线时应引起重视。L.brevisP3发酵液中苯乳酸测定结果为(0.1400±0.0046) mg/L。
2.4 乳酸菌发酵液中苯乳酸的测定
本实验取L.brevisP3和L.plantarumATCC8014发酵液检测其中苯乳酸的含量。取灭菌后的MRS液体培养基测定空白样品,检测发现MRS液体培养基含有苯乳酸,含量为5.58 mg/L。乳酸菌发酵液中苯乳酸含量为测定值减空白样品中苯乳酸含量。
如图3所示,L.plantarumATCC8014和L.brevisP3均能够合成苯乳酸,在培养120 h后苯乳酸合成量分别为77.58和62.60 mg/L。利用本方法,苯乳酸在6.00 min出峰,目标峰与杂质峰分离彻底,出峰时间适宜,检测成本低,有机相使用比例小,环境友好。本方法可根据待测物质中苯乳酸的含量调整标准曲线的线性范围。

图3 乳酸菌发酵液中苯乳酸HPLC图谱
3 讨论
C18和C8表示通过固定相键合至二氧化硅的烷烃分别为18或8个碳。碳链的增加也增加了键合相的非极性区域,能够影响保留和选择性[24]。C18的碳链比C8长,因此具有更好的保留性能。C8色谱柱适用于大分子的分离,而苯乳酸是小分子物质,C18色谱柱可以更好地满足检测需求[24]。色谱柱封端主要用于反相色谱中。封端色谱柱由于覆盖了更多的硅烷醇基,可以消除或减少可能发生的副反应,减少拖尾并延长色谱柱的寿命。由于发酵液和水相流动相为酸性,使用色谱柱II可以减少色谱柱堵塞的可能性,从而延长色谱柱的使用寿命。故选择封端C18色谱柱为佳。
采用酸性的水相流动相能够改善峰型,苯乳酸为小分子有机酸,采用酸性的流动相也能够抑制待测物质解离[25-27]。水相流动相使用前测定pH,保证pH>2,防止酸性过大时容易对色谱柱造成损伤。TFA的最大吸收波长小于200 nm,苯乳酸的检测波长为210 nm,检测干扰小。由于苯乳酸RP-HPLC中的保留值较小,测定时一般采用极性大的流动相,当酸性流动相比例高时,酸性物质容易被电离,导致出现小峰,故苯乳酸在0.1%甲酸-乙腈和乙酸铵-乙腈下出现小峰和峰型分裂[26]。0.1%甲酸水溶液、0.02 mol/L KH2PO4溶液和0.02 mol/L K2HPO4和乙腈在75∶25 (v/v)下轻微拖尾,峰高一致,不影响定性定量,但当样品中杂质过多时,容易造成苯乳酸目标峰和杂质峰分离不开,检测结果误差大,故选择0.05%TFA-乙腈为流动相,峰型对称,半峰宽小,定性定量更加准确。
目前苯乳酸的HPLC检测多采用梯度洗脱,梯度洗脱可用于一次分离多种物质,当分离物质只有一种时,梯度洗脱容易造成基线不稳,影响定型和定量,且梯度洗脱所需要的平衡时间长,采用等度洗脱的方法,平衡所需的时间短,也就缩短了测定所需时间,随着色谱柱平衡时间的减少和有机试剂的使用减少,节省了检测成本,对环境友好,不仅能够应用于实验室内苯乳酸含量的测定,还能够用于大批量样品的测定。利用本方法检测乳酸菌发酵液中的苯乳酸,出峰稳定,目标峰与杂质峰分离程度好,操作简单,定性定量准确。
李兴峰等[28-29]从家庭中式泡菜中分离出能够合成苯乳酸的L.plantarum和干酪乳杆菌(Lactobacilluscasei),市售泡菜中没有分离到乳酸菌。L.brevisP3分离自市售中式泡菜,具有吸附重金属Cr3+的能力[30]。L.brevisP3不仅能够吸附重金属,还具有合成苯乳酸的能力,对重金属吸附和苯乳酸合成的综合性研究,具有一定的参考性价值,L.plantarumATCC8014能够在120 h内合成77.58 mg/L苯乳酸,可在后续的研究中对其苯乳酸合成能力进行优化。
4 结论
本实验对一种RP-HPLC检测乳酸菌发酵液中苯乳酸的方法从流动相和固定相进行了优化,对能够用于苯乳酸检测的流动相和固定相进行了测试和评价,确定了使用封端C18色谱柱,0.05%TFA:乙腈,体积比为75∶25的等度洗脱的洗脱系统,检出限和定量限分别为0.1400和0.4600 mg/L,仪器精密度良好,回收率(99.88%~101.08%)较高,方法重复性能够满足检测需要,操作简单,结果准确,可以用于检测发酵液中苯乳酸,发现了两株能够合成苯乳酸的乳酸菌,L.plantarumATCC8014和L.brevisP3均能够合成苯乳酸,在培养120 h后苯乳酸合成量分别为77.58和62.60 mg/L,为乳酸菌合成苯乳酸的相关研究提供参考,后续研究中可对两株乳酸菌菌合成苯乳酸能力进一步考察和优化。
