气相色谱法测定糖精钠中邻、对甲苯磺酰胺含量
2020-07-04张东阁郭艳玲宇杰
张东阁 郭艳玲 宇杰

[摘要]目的 建立气相色谱法(GC)测定糖精钠中邻、对甲苯磺酰胺。方法 DB-17MS毛细管色谱柱,初始温度180℃保持20 min,以10℃/min升至260℃,保持32 min。结果 邻甲苯磺酰胺在11.80~223.60 μg/ml(r=1)内峰面积与浓度呈良好线性关系,平均加样回收率为96.58%,RSD为5.73%(n=6)。对甲苯磺酰胺在10.15~203.00 μg/ml(r=1)内峰面积与浓度呈良好线性关系,平均加样回收率为103.99%,RSD为2.83%(n=6)。6个批次的糖精钠中邻、对甲苯磺酰胺含量测定由于结果低于检测限,未检出,符合药典要求。结论 GC测定糖精钠中甲苯磺酰胺含量操作简便、准确,可作为糖精钠的质量评价标准。
[关键词]气相色谱法;糖精钠;甲苯磺酰胺;含量测定
[中图分类号] R917 [文献标识码] A [文章编号] 1674-4721(2020)5(c)-0031-05
Content determination of o/p-toluenesulfonamide in saccharin sodium by the gas chromatography
ZHANG Dong-ge1,2 GUO Yan-ling2 YANG Yu-jie1
1. Chengde Medical University, Hebei Province, Chengde 067000, China; 2. Jingfukang Pharmaceutical Group Co., Ltd., Hebei Province, Chengde 067000, China
[Abstract] Objective To establish the gas chromatography for the determination of o/p-toluenesulfonamide in saccharin sodium. Methods DB-17MS chromatographic column was applied, the initial temperature was set at 180 ℃ for 20 min, and then heated up to 260℃ at 10℃/ min and maintained for 32 min. Results The peak area of o-toluenesulfonamide in 11.80-223.60 μg/ml (r=1) showed a good linear relationship with the concentration, the average sample recovery rate was 96.58%, and the RSD was 5.73% (n=6). The peak area of p-toluenesulfonamide in 10.15-203.00 μg/ml (r=1) showed a good linear relationship with the concentration, the average sample recovery rate was 103.99%, and the RSD was 2.83% (n=6). The content of o/p-toluenesulfonamide in saccharin sodium in 6 batches was not detected because the results were lower than the detection limit, which met the requirements of the pharmacopoeia. Conclusion The GC for the determination of toluenesulfonamide in saccharin sodium is simple and accurate, and can be used as a quality evaluation standard for saccharin sodium.
[Key words] Gas chromatography; Saccharin sodium; Toluenesulfonamide; Content determination
糖精鈉是一种常用的甜味剂,据报道,在美国广泛应用于食品和医药行业[1]。糖精钠甜度是蔗糖的300~500倍,除了在味觉引起甜的感觉外,对人体无任何营养价值,安全性至今存在较大争议[2-3]。糖精钠利用甲苯法生产过程中,一般会生成甲苯磺酰胺这种中间品,由于生产过程中氧化不彻底,未完全反应的甲苯磺酰胺未完全分离,而甲苯磺酰胺属于致癌物[1]。甲苯磺酰胺包括邻甲苯磺酰胺和对甲苯磺酰胺,两种成分为同分异构体。另有文献报道糖精钠中邻、对甲苯磺酰胺通常认为有致癌作用,所以样品中这两种杂质的测试十分重要[4]。国内外标准均控制糖精钠中杂质邻、对甲苯磺酰胺的含量[5]。联合国粮农组织和世界卫生组织食品附加剂联合专家委员会第十八次会议决定限制糖精钠中全部甲苯磺酰胺异构体少于100 PPM[6]。2015版《中华人民共和国药典》(以下简称《中国药典》)[7]对糖精钠中邻、对甲苯磺酰胺含量进行了控制,采用的是气相色谱法(gas chromatography,GC),但在实际检验过程中存在色谱条件分离度不佳的情况。另有研究人员采用紫外分光光度法和高效液相色谱法测定邻、对甲苯磺酰胺含量[8]。但是经分析紫外分光法,准确度不高;高效液相色谱法分析时间较长。目前国内利用GC同时测定邻、对甲苯磺酰胺含量报道较少。本研究在《中国药典》[7]方法的基础上对色谱条件进行了改进和优化,以期得到一种更可行的色谱方法。
1仪器、试剂与试药
1.1仪器与试剂
GC仪(岛津,GC-2010);色谱柱:DB-17MS(30 m×0.25 mm×0.25 μm);FID检测器;硅藻土(分析纯,天津欧博凯化工有限公司)、盐酸(分析纯,天津欧博凯化工有限公司)、二氯甲烷(优级纯,天津欧博凯化工有限公司);玻璃色谱管(250 mm×25 mm)。
1.2试药
邻甲苯磺酰胺对照品(中国食品药品检定研究院,100038-201204);对甲苯磺酰胺(中国食品药品检定研究院,100131-201603);糖精钠(湖北华纳大药厂股份有限公司,批号:171101、171102、171108、171114、171105、171109、171111)。
2方法与结果
2.1气相色谱系统适应性条件
GC仪;色谱柱:DB-17MS;采用程序升温:初始温度180℃保持20 min,以10℃/min升至260℃,保持32 min;进样量:1 μl;柱流速:1.5 ml/min;分流比:5∶1;进样口温度:200℃;检测器温度:280℃。
2.2溶液制备
2.2.1混合对照品溶液的制备 分别取邻甲苯磺酰胺与对甲苯磺酰胺对照品,精密称定(邻甲苯磺酰胺称样量:0.012 53 g;对甲苯磺酰胺称样量:0.012 56 g),加二氯甲烷溶解并稀释制成每1毫升含上述的甲苯磺酰胺异构体各50 μg的溶液作为对照品混合溶液。
2.2.2供试品溶液的制備 供试品溶液的制备:取本品2.0 g(称样量1:2.002 g;称样量2:2.004 g),精密称定,用5%碳酸钠溶液8.0 ml溶解后,加色谱用硅藻土[称取硅藻土(过九号筛)100 g,加盐酸800 ml,时时搅拌,浸渍12 h以上,除去酸液,再用盐酸同样处理3次,每次1 h,然后用水洗涤至溶液显中性,将硅藻土分散于甲醇300 ml中,滤过,在80℃烘干]10 g,混合均匀,装入25 mm×250 mm的色谱管,照柱色谱(通则0511第二法),用二氯甲烷洗脱约30 min,收集洗脱液50 ml,蒸发至近干,加二氯甲烷,使成1.0 ml作为供试品溶液。
2.3标准曲线制备
取邻甲苯磺酰胺对照品与对甲苯磺酰胺对照品,精密称定分别置10 ml容量瓶中,加二氯甲烷溶解并稀释至刻度,分别制成每1毫升含上述的甲苯磺酰胺异构体各1 mg的对照品储备液。经称量后最终邻甲苯磺酰胺储备液1.118 mg/ml,对甲苯磺酰胺储备液1.015 mg/ml。
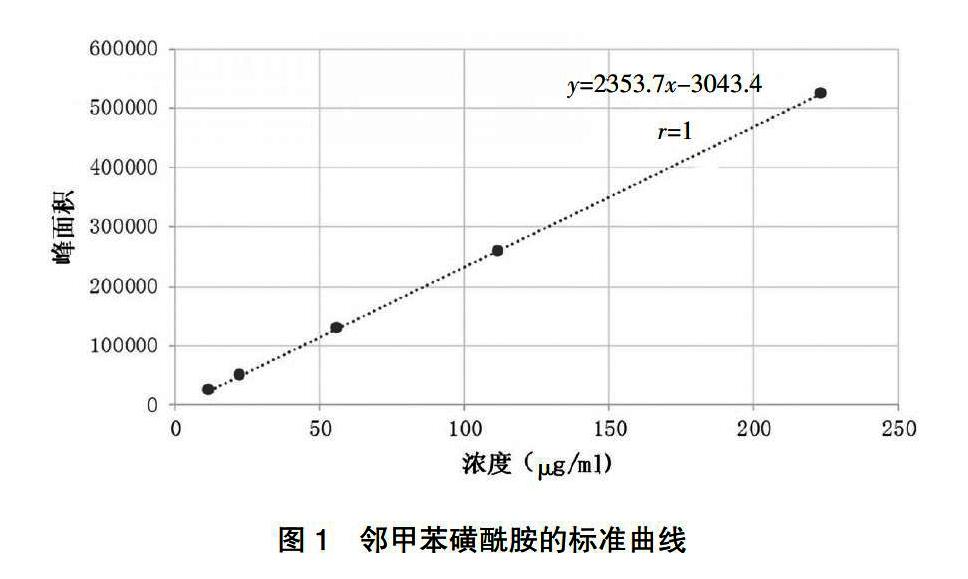
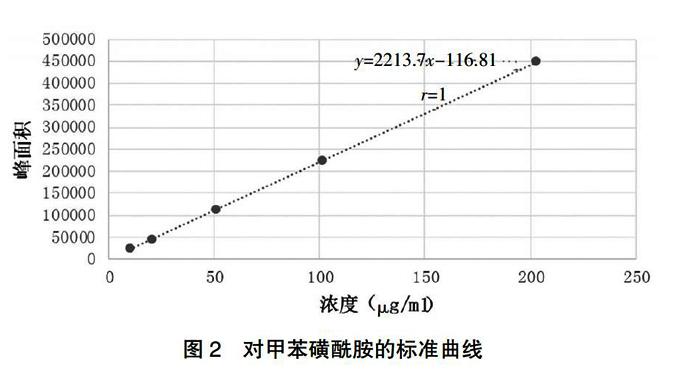
分别精密吸取两种储备液0.1、0.2、0.5、1、2 ml置5个10 ml容量瓶中,用二氯甲烷稀释至刻度,摇匀,即得系列浓度的邻甲苯磺酰胺与系列浓度的对甲苯磺酰胺混合对照品溶液。进GC样,记录峰面积。结果显示,邻甲苯磺酰胺线性回归方程为y=2353.7x-3043.4,邻甲苯磺酰胺浓度在11.80~223.60 μg/ml内线性关系良好(r=1);对甲苯磺酰胺线性回归方程为y=2213.7x-116.81,对甲苯磺酰胺浓度在10.15~203.00 μg/ml内线性关系良好(r=1)(表1,图1~2)。
2.4检出限
参考文献[9-13]了解检出限的具体细节后,取邻甲苯磺酰胺对照品与对甲苯磺酰胺对照品约10 mg,精密称定分别置25 ml容量瓶中,加二氯甲烷溶解并定容至刻度,制得两种对照品储备液。经称量定容后得:邻甲苯磺酰胺储备液浓度为427.6 μg/ml,对甲苯磺酰胺储备液浓度为431.2 μg/ml。
分别精密吸取两种同分异构体对照品储备液各0.2 ml,置两个不同5 ml容量瓶中,加二氯甲烷稀释至刻度,即得邻甲苯磺酰胺母液(17.104 μg/ml)、对甲苯磺酰胺母液(17.248 μg/ml)。再分别精密吸取两种母液各0.2 ml置同一10 ml容量瓶中,加二氯甲烷稀释至刻度,即得甲苯磺酰胺异构体检出限浓度。邻甲苯磺酰胺检出限浓度为0.342 08 μg/ml,对甲苯磺酰胺检出限浓度为0.344 96 μg/ml。邻甲苯磺酰胺信噪比为3.33,对甲苯磺酰胺信噪比为2.04,全部符合2015版《中国药典》[7]要求。
2.5仪器精密度
取邻甲苯磺酰胺与对甲苯磺酰胺对照品,精密称定,加二氯甲烷溶解并稀释制成每1毫升含上述的甲苯磺酰胺异构体各50 μg的溶液作为对照品溶液,具体如下。
分别精密吸取邻甲苯磺酰胺储备液(427.6 μg/ml)、对甲苯磺酰胺储备液(431.2 μg/ml)1.2 ml,置同一10 ml容量瓶中,加二氯甲烷稀释至刻度即得。进GC样,记录峰面积。邻甲苯磺酰胺:51.312 μg/ml,对甲苯磺酰胺:51.744 μg/ml。精密度结果具体见表2。
2.6定量限
参考文献[14-17]了解定量限的细节后,取邻甲苯磺酰胺对照品与对甲苯磺酰胺对照品约10 mg,精密称定分别置25 ml容量瓶中,加二氯甲烷溶解并稀释至刻度,制得两种储备液。邻甲苯磺酰胺储备液浓度:427.6 μg/ml,对甲苯磺酰胺储备液浓度:431.2 μg/ml。
分别精密吸取两种储备液各0.2 ml,置两个不同5 ml容量瓶中,即得邻甲苯磺酰胺母液(17.104 μg/ml)、对甲苯磺酰胺母液(17.248 μg/ml)。再分别精密吸取邻甲苯磺酰胺0.6 ml,对甲苯磺酰胺0.9 ml置同一10 ml容量瓶中,加二氯甲烷稀释至刻度,即得甲苯磺酰胺异构体定量限浓度。邻甲苯磺酰胺定量限浓度:1.026 24 μg/ml,对甲苯磺酰胺定量限浓度:1.552 32 μg/ml。邻甲苯磺酰胺信噪比为10.86,对甲苯磺酰胺信噪比为10.67,全部符合2015版《中国药典》[7]要求。
2.7稳定性实验
取本品1.0 g,精密称定,精密加入混合对照品溶液0.5 ml(约25 μg)用5%碳酸钠溶液8.0 ml溶解后,加色谱用硅藻土[称取硅藻土(过九号筛)100 g,加盐酸800 ml,时时搅拌,浸渍12 h以上,除去酸液,再用盐酸同样处理3次,每次1 h,然后用水洗涤至溶液显中性,将硅藻土分散于甲醇300 ml中,滤过,在80℃烘干]10 g,混合均匀,装入25 mm×250 mm的色谱管,照柱色谱(通则0511第二法),用二氯甲烷洗脱约30 min,收集洗脱液50 ml,蒸发至近干,加二氯甲烷,使成1.0 ml作为供试品溶液。分别于0、1、2、4、6、8、12 h进GC样,记录峰面积,计算RSD,考察稳定性,具体结果见表3。
2.8重复性实验
按“2.2.1项”下制备混合对照品溶液,同时精密称定2.0 g糖精钠,按“2.2.2项”下制备6份供试品溶液,进GC样,记录峰面积,计算含量,具体结果见表4。
2.9回收率实验
2.9.1对照品储备液制备 取邻甲苯磺酰胺对照品与对甲苯磺酰胺对照品,精密称定分别置10 ml容量瓶中,加二氯甲烷溶解并稀释至刻度,分别制成每1毫升含上述的甲苯磺酰胺异构体各1 mg的对照品储备液。邻甲苯磺酰胺贮备液浓度:1.119 mg/ml,对甲苯磺酰胺贮备液浓度:1.097 mg/ml。
2.9.2混合对照品溶液的制备 分别精密吸取甲苯磺酰胺异构体贮备液各0.5 ml,置同一10 ml容量瓶中,加二氯甲烷稀釋至刻度,即得。邻甲苯磺酰胺浓度:55.95 μg/ml,对甲苯磺酰胺浓度:54.85 μg/ml。
2.9.3加样回收样品制备 取本品1.0 g,精密称定,精密加入混合对照品溶液0.5 ml(约25 μg)用5%碳酸钠溶液8.0 ml溶解后,加色谱用硅藻土[称取硅藻土(过九号筛)100 g,加盐酸800 ml,时时搅拌,浸渍12 h以上,除去酸液,再用盐酸同样处理3次,每次1 h,然后用水洗涤至溶液显中性,将硅藻土分散于甲醇300 ml中,滤过,在80℃烘干]10 g,混合均匀,装入25 mm×250 mm的色谱管,照柱色谱(通则0511第二法),用二氯甲烷洗脱约30 min,收集洗脱液50 ml,蒸发至近干,加二氯甲烷,使成1.0 ml作为供试品溶液。结果显示,随行含测未检出,加样回收实验结果具体见表5。
2.10样品含量测定
取6批样品,按“2.2.2项”下方法制备样品,同时按“2.2.1项”下方法制备混合对照品溶液,进GC样,即得,具体结果见表6。
《中国药典》[7]规定糖精钠中总的甲苯磺酰胺 <0.0025%,6批样品未检出符合药典规定,同时也与生产厂家的报告单一致。
3讨论
本研究中,样品检测时未检出邻、对甲苯磺酰胺,这与检验报告单数据一致。分析原因可能为糖精钠在生产过程中产生邻、对甲苯磺酰胺中间品,而在后期的氧化过程中,两种中间品大部分被分离,仅剩余非常少的一部分,而剩余的少部分低于检出限,很可能埋于噪音中,所以本研究不能绝对地断定样品中一点也不含邻、对甲苯磺酰胺。本次方法学考察由于预实验时样品中未检出甲苯磺酰胺,故在考察样品的稳定性时利用加混合对照方法。在方法学考察过程中,两种同分异构体检出限浓度和定量限浓度确定过程中要首先做预实验,在预实验得出浓度的基础上再逐级稀释直至符合限度要求。2015版《中国药典》[7]要求邻、对甲苯磺酰胺之和不得超过0.0025%,根据药典规定本数量级RSD要求不高于6.0%[7],故本研究实验方法学的回收率实验RSD数据合理。该方法的分离度良好,邻、对甲苯磺酰胺定量检测的线性范围广,灵敏度高,并且具有良好的精密度,可以用于邻、对甲苯磺酰胺的检测。本次实验过程中所用到的中性硅藻土提取过程复杂,盐酸使用量大,在制备过程中盐酸需注意安全并及时回收避免污染环境。
生产中要求快速准确测定出对甲苯磺酰胺和邻甲苯磺酰胺含量,测定方法包括凝固点法与紫外光谱法[18-21],但两种方法要求操作条件比较苛刻,不易操作成功,而相关文献[4,6]虽然采用的也是气相色谱分析方法,但是两种方法在方法学考察过程过于简单,且并未做评价方法准确度的加样回收实验,本研究建立的GC色谱条件分离度较好,方法操作简便,准确,可作为糖精钠的质量评价标准参考,具有一定的推广价值。
[参考文献]
[1]刘娟娟,舒静,张莉.糖精钠行业存在问题和解决措施[J].价值工程,2015,34(7):305-306.
[2]钱晓翠,黄平,周艳霞.HPLC法测定铝碳酸镁混悬液中苯甲酸钠和糖精钠的含量[J].中国合理用药探索,2019,16(5):179-182.
[3]曹张欢.两种国家标准方法在HPLC法测定白酒中糖精钠检出限的比较[J].食品安全导刊,2019,(21):123.
[4]宋子台,陈顺喜,李凡庆.紫外分光光度法测定糖精钠中微量邻、对甲苯磺酰胺[J].分析测试通报,1986,5(3):37-39.
[5]李凤新.GC法测定邻、对甲苯磺酰胺[J].科技创新与应用,2013,4(21):43.
[6]周琢.糖精中邻苯磺酰胺的测定[J].河南预防医学杂志,1976,1(16):76-77.
[7]国家药典委员会.中华人民共和国药典[S].二部.北京:中国医药科技出版社,2015:1562.
[8]徐秀珠,张骅,傅小芸,胡耿源.紫外光谱法和高效液相色谱法测定邻、对甲苯磺酰胺[J].分析化学,1992,(12):1455-1457.
[9]徐建白,唐云,庞东颖,等.HPLC法测定活络酊中水杨酸甲酯[J].中草药,2011,42(10):2026-2027.
[10]夏红兰,姜燕燕,石任兵.百合知母汤有效部位质量控制方法研究[J].中国实验方剂学杂志,2013,19(4):126-130.
[11]隋继英,刘向红,陈金娜,等.化瘀无糖颗粒的质量标准研究[J].中国药房,2018,29(22):3083-3088.
[12]刘栋,李妍.探讨分析化学中几项分析性能指标[J].广州化工,2019,47(8):137-138,172
[13]杨淳,郑珊,刘洋,等.分析化学中“检出限”的物理意义及测定方法探究[J].大学化工,2018,33(5):51-55.
[14]关小娟,吴查清.顶空气相色谱法测定强力枇杷露中薄荷脑的含量[J].中国药房,2016,27(12):1708-1710.
[15]黄艳,陈雪云,杨雪峰,等.高效液相色谱法测定利伐沙班中的有关物质[J].中国当代医药,2017,24(24):78-81.
[16]袁彩君,朱天新.高效液相色谱法測定左卡尼汀注射液的含量及杂质A[J].中国当代医药,2015,22(19):8-12,20.
[17]万妮,孙园媛,吴广野.气相色谱法对西葫芦中农药多残留定量限的测定[J].农业科技与装备,2012,23(7):26-28.
[18]陈飙,胡国忠.GC法测定邻、对甲苯磺酰胺[J].辽宁化工,1996,(1):59-60.
[19]陈立光,梁明泉,邓庆荣,等.采用气相色谱法建立氟[18F]脱氧葡糖注射液中有机溶剂残留的测定方法及其方法不确定度的评价[J].中国医药科学,2018,8(9):49-52.
[20]杨梅,欧嘉娜.高效液相色谱-示差折光检测器法测定聚多卡醇注射液的有关物质和含量[J].中国当代医药,2019, 26(22):15-18.
[21]袁惠霞,欧国灯,刘潇潇.气相色谱法测定香砂六君丸中乙酸龙脑酯[J].中国医药科学,2018,8(19):69-71.
(收稿日期:2019-09-16 本文编辑:任秀兰)
[作者简介]张东阁(1988-),男,内蒙古赤峰人,承德医学院2019级在读硕士研究生,主要从事中药新药研究与开发
通讯作者:杨宇杰,女,教授,硕士研究生导师,主要从事中药新药研究与开发
