X-连锁隐性遗传Charcot-Marie-Tooth 病5 型1 例报告并文献复习
2020-06-30夏静宜
徐 敏 夏静宜 郭 虎
南京医科大学附属儿童医院神经内科(江苏南京 210008)
Charcot-Marie-Tooth病(Charcot-Marie-Tooth disease,CMT)也称腓骨肌萎缩症,由Jean Charcot、Pierre Marie及Howard Henry Tooth于1886年首次描述,是一组慢性进行性周围神经病,以四肢远端肌肉无力和萎缩为主要表现,可伴有感觉和植物神经症状。CMT具有多种不同的致病基因和遗传方式,其中X-连锁隐性遗传CMT(X-linked Charcot-Marie-Tooth disease,CMTX)又进一步分为CMTX 1 到CMTX 6亚型[1]。本文回顾分析1 例PRPS 1基因突变导致的CMTX 5 型(CMTX 5)患儿的临床资料,并进行文献复习,以期提高对CMTX5的认识。
1 临床资料
患儿,男,14 岁5 个月。自幼听力丧失,3 岁时安装人工耳蜗,6岁发现行走无力,并进行性加重,步态异常,跛行,两年前外院行跟腱松解延长术,术后步态稍好转。智力语言发育无异常,学习成绩中等偏下。患儿系G1P1,足月顺产,父母体健,非近亲婚配,家族史无异常。体格检查:神清,反应可,双侧眼球各方向活动正常,心、肺、腹无异常,双上肢肌力无异常,双下肢近端肌力4 级,远端肌力3~4 级,双上肢肌张力正常,双下肢肌张力下降。马蹄内翻足,高足弓,左侧著,双足趾屈曲挛缩,主被动活动受限,踝关节屈曲挛缩,主被动背伸活动受限,屈曲尚可,跟腱挛缩,跨域步态,双侧膝腱反射均减弱,双下肢温痛觉减退。肌电图:受检运动神经复合肌肉动作电位(compound muscle action potential,CMAP)波幅均降低或未引出,伴运动神经传导速度(motor nerve conduction velocity,MCV)减慢(≥3 SD),伴或不伴远端潜伏期轻度延长(>2~3 SD),双侧胫运动神经F波及H反射均未引出,右侧正中、尺神经最短潜伏期延长,伴或不伴出波率降低;被检感觉神经动作电位(sensory nerve action potential,SNAP)波均未引出;提示多发性周围神经源性损害肌电改变(主要累及感觉、运动神经轴索损害伴脱髓鞘);视觉诱发电位未见异常;双眼视力及眼底检查无异常。
为明确诊断,经患儿监护人知情同意,采集患儿及其父母外周血标本各2 mL,送至凯昂医学检验所进行全外显子检测,并采用Sanger测序法进行位点验证及家系验证。结果显示:患儿PRPS1基因存在c.344T>C错义变异;Sanger测序提示父亲无变异,母亲为杂合变异,变异来源于母亲(图1)。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南评定为疑似致病性变异。该致病位点既往已有报道。根据患儿耳聋、肌无力症状,肌电图改变及基因检测结果,最终诊断为CMTX5。
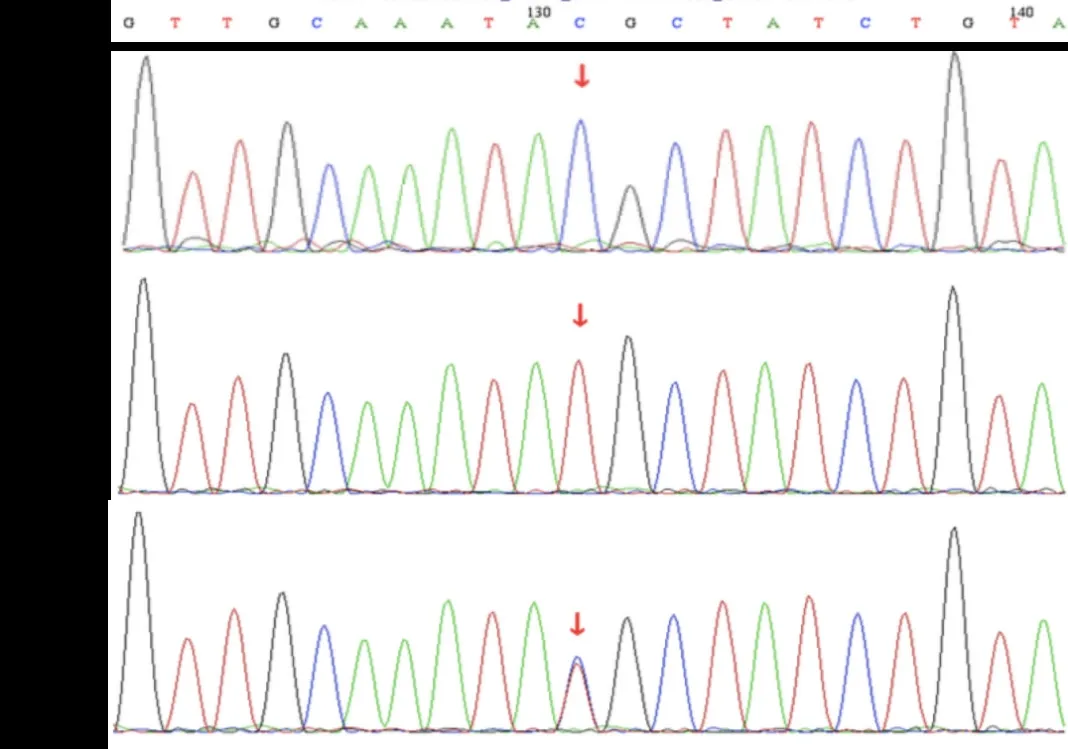
图1 患儿及父母PRPS1 基因序图
2 讨论
CMTX 5 由PRPS 1基因变异引起。PRPS 1基因突变可导致磷酸核糖焦磷酸酶(phosphoribosyl pyrophosphate synthetase I,PRS-1)活性增加或减少。PRPS 1缺陷分为过表达、严重缺陷、中度及轻度缺陷4个类型,分别导致PRS-1超活性,Arts综合征,CMTX5和非综合征性感音神经性耳聋(nonsyndromic sensorineural deafness,DFN2),这4种疾病均为X连锁隐性遗传[2],且各型之间有一定的交叉重叠,是PRPS1基因突变导致的疾病谱[3]。PRS-1是核苷酸合成的初级阶段的必需酶,具有六聚体结构,也是嘌呤,嘧啶和吡啶核苷酸合成的必需辅助因子[2]。PRPS1变异影响重要的细胞功能,包括核酸合成,能量代谢和细胞信号传导。PRPS1相关神经功能障碍可能与神经元脱髓鞘,嘧啶合成受损,三磷酸鸟苷水平降低以及吡啶核苷酸合成的丧失有关。而神经元细胞具有高能量需求,当三磷酸鸟苷(GTP)和其他嘌呤核苷酸(包括ATP)的水平降低时,神经、肌肉和视网膜等能量供应不足,从而引起一系列相关临床症状[2,3]。
CMTX 5 以早期感觉神经性耳聋,周围神经病变和视神经病变为典型三联征。CMTX 5 的男孩患有早发性(语前)感觉神经性听力损失,部分患儿生后即听力丧失。周围神经病变出现的年龄通常为5~12岁,最初表现是行走无力,步态异常,腱反射减弱或消失,以运动神经受累明显,肌肉无力一般下肢早于上肢,且更为严重。随着疾病的进展可出现感觉神经损害,出现感觉异常,包括麻木、灼烧和刺痛感,或手脚感觉丧失;也可合并骨骼异常,如锤头趾畸形和高足弓[4]。视力障碍出现的年龄为7~20岁。周围神经病变和视力障碍都随着时间推移而进展,严重者最终可能会失明,行动需要依赖拐杖或轮椅,但一般不影响智力和寿命。除了典型的三联征,有的患儿在病程初期仅表现为发热后一过性肌无力,感染后1个月可恢复至病前状态[5];也有患者出现共济失调,精神行为障碍,表现为注意力不集中,冲动和攻击行为等[6]。女性携带者也可出现轻度的临床症状,如不同程度的听力损害[6]。对这部分临床表现不典型的患者,需提高警惕。
迄今为止,基因型与表型的相关性尚不明确。以“腓骨肌萎缩症”、“Charcot-Marie-Tooth disease”、“PRPS1”为关键词,对中国期刊全文数据库(CNKI)、万方数据知识服务平台和美国国家生物技术中心(NCBI)、生物医学文献数据库(PubMed)数据库建库至2019年8月收录的文献进行检索,共检索到6篇相关报道(均为英文文献)。已报道的引起CMTX5的PRPS1基因型仅有8种,突变主要在第3外显子,分别为c.344T>C、c.129A>C[7],c.362C>G[8],c.830A>C[6],c.343A>G、c.925G>T[9],c.319A>G[5]和c.202A>T[10]。文献报道的8种基因型,听力损害和周围神经病是共同的表现,而视觉障碍具有个体差异。本例及文献报道9 例患者中有4 例出现视觉障碍。本例患儿就诊时已14岁,尚未出现视力障碍。有报道15岁CMTX5患儿(基因型c.362C>G)没有出现视力障碍[8],但也有报道10岁患儿(c.344T>C)已出现视觉损害[7],提示CMTX 5 临床表型不仅仅与PRPS 1变异位点有关,也与其他因素有关,比如残存的PRS-1酶活性。在一个CMTX5的德国家系中(基因型c.830A>C),男性患者未检测到PRS-1酶活性,而在有症状的基因携带者姐妹中活性减少,在无症状携带者中活性无异常;即使相同基因型的女性携带者,酶的活性也因人而异,推测可能与X染色体失活倾斜程度有关[6]。因此,PRS-1酶残留活性检测有助于确立临床表型,尽早把握患者的病程进展。总之,PRPS1变异位点和残留酶活性可能是临床表型的主要决定因素,其他影响因素还有待进一步研究,也需要更大的CMTX5样本量累积。
目前,CMTX 5 尚无根治方法,康复训练、适当的外科手术以及必要的社会心理治疗能一定程度改善症状[11]。基因靶向治疗也处于研究阶段。研究发现,腺苷甲硫氨酸补充剂有助于提高ATP和GTP浓度,延缓听力损害并减轻神经症状[12]。虽然无特效治疗手段,早期明确诊断也有助于家庭的遗传咨询和产前诊断,改善患儿生活质量。
综上,即使CMTX5的基因型相同,临床表型也不尽相同,表型与基因型相关性需进一步大样本研究。
