间作不同农作物对刺梨园土壤微生物类群及酶活性的影响
2020-05-27刘岚君何季文雪峰
刘岚君 何季 文雪峰


摘要:为探究间作不同农作物对刺梨园土壤微生物类群和酶活性的影响,在贵州省黔南州龙里县谷脚镇刺梨种植基地设置刺梨/辣椒间作、刺梨/玉米间作和刺梨单作三个种植模式的试验区,取0~20 cm深度和20~40 cm深度的土样,分别测试其土壤微生物数量和酶活性,探明不同种植模式下土壤微生物(细菌、真菌、放线菌)数量和酶活性的变化。结果表明:间作对刺梨园土壤微生物数量和酶活性有促进的作用。间作模式下土壤微生物数量明显高于单作,综合比较发现:玉米//刺梨>辣椒//刺梨>刺梨单作;间作模式下土壤酶活性高于刺梨单作,其中:玉米//刺梨>辣椒//刺梨>刺梨。分析还表明:刺梨园土壤微生物数量和土壤酶活性之间呈正相关关系。
关键词:刺梨园土壤;间作;微生物;酶活性
中圖分类号:S1542;S1543
文献标识码:A
文章编号:1008-0457(2019)06-0008-06国际DOI编码:10.15958/j.cnki.sdnyswxb.2019.06.002
Effects of Intercropping Different Crops on Soil Microbial Group and Enzyme Activities in Rose Roxburghii Orchard
LIU Lan-jun,HE Ji*,WEN Xue-feng
(College of Agriculture,Guizhou University,Guiyang,Guizhou 550025,China)
Abstract:In order to explore the effects of intercropping different crops on soil microbial groups and enzyme activities in Rose roxburghit,a test area with three planting patterns:the thorn pear/pepper intercropping,thorn pear/maize intercropping and thorn pear monocropping in the thorn pear planting base in Gujiao Town,Longli County,Guizhou Provincesoil samples with depths of 0~20 cm and depths of 20~40 cm were tested for soil microbial quantity and the changes of soil microbes (bacteria,fungi,actinomycetes)and enzyme activities under different planting patterns The results showed that intercropping had a promoting effect on soil microbial quantity and enzyme activity in the thorn garden The amount of soil microbes in the intercropping mode was significantly higher than that in the single crop The comprehensive comparison showed that:corn-thorn pear> pepper-thorn pear> thorn pear single crop; soil enzyme activity in the intercropping mode was higher than that of thorn pear in the single crop,among which:corn-thorn pear >pepper-prickly pear > prickly pear single The analysis also showed that there was a positive correlation between soil microbial quantity and soil enzyme activity in the garden
Key words:Rose roxburghii orchard soil; intercropping; microorganism; enzyme activity
间作是指在同一块土地上,分行或者分带间种植2种或2种以上生长周期相同或相近的作物,该种植模式可以根据它的特性来充分地利用土地[1]。合理的间作模式具有密植效应、时空效应、补偿效应、边际效应、异质效应,提高了对土地、养分、光能、热能的利用率[2-3],实现农业的高产高效。间作的种植模式除了提高作物的产量,还能提高作物的抗病率。间作的种植模式使经济和生态的双赢[4]。间作对土壤微生物的数量和酶活性有着积极的影响,张向前、黄国勤等人[5]研究发现,和单作相比,玉米和花生间作可以促进或提高土壤微生物的群落功能多样性和土壤酶活性,并且对玉米的产量和品质有着显著的提高作用。微生物是土壤的重要组成部分,土壤酶是一种生物活性物质,主要来自于植物根系、土壤微生物和动植物残体分解[6-7]。它们都参与土壤的生化反应,推动了土壤生理生化反应的进程,是土壤生态中不可缺少的对象[8],因而研究土壤微生物和酶,对评价土壤质量和肥力有重要意义。微生物对土壤的微环境十分敏感,受很多因素的影响。就栽培方面而言,施肥、嫁接、轮作、间作等对微生物的数量有显著影响。前人的研究表明,间作的种植模式相对于单作能提高土壤微生物的根际效应,提高土壤微生物的含量,改善土壤的微环境,进而改善土壤的酶活性[9-14]。土壤中微生物所分泌的某些化学物质能够对酶形成及其活性产生重要影响,因此,土壤中细菌、真菌、放线菌的数量与酶的数量和活性有着非常紧密的关系。
刺梨富含维生素C,对人类身体有益。通过间作方式种植刺梨,不仅可以助力脱贫攻坚,而且还可以提高土地的利用率。本文探讨不同间作模式对刺梨园土壤微生物类群(细菌、真菌、放线菌)的数量和土壤酶活性的影响,进而分析间作与单作的种植方式下土壤肥力的状况,为农民提供刺梨间作种植的生态管理依据。
1材料与方法
11研究区概况
本试验设在贵州省黔南州龙里县谷脚镇刺梨种植基地,地处东经106°45′19″~107°15′1″,北纬26°10′19″~26°49′33″之间,海拔介于1080~1500 m之间。该区属亚热带季风湿润气候,年降雨量平均为10893 mm,平均气温15℃。喀斯特地貌明顯,黄壤。
12材料与方法
121样地设置及样品采集
根据典型性原则和代表性原则在研究区设置两种不同间作模式及单种刺梨的对照试验区(见表1)。共三个处理,每个处理三次重复,共9个实验小区,试验区为平地,每个样地的面积为15 m×15 m,种植刺梨的品种为贵农5号,施用的肥料统一为氮磷钾复合肥。
2017年1~2月期间对每个样地0~20 cm和20~40 cm土层分层采集土样,采用“S形”取样,四分法混匀样品,每个样品取1~15 kg。取回的土样部分放在冰箱冰藏,用于测定土壤微生物。部分土样风干研磨后过1 mm筛,装袋储藏用来测定土壤酶。
122测定方法
土壤微生物的测定方法:首先称取10 g土壤样品放入盛有90 mL无菌水的三角瓶中,置于摇床上振荡1 h,静置30 min,使土样与水分充分混匀,使土壤中的微生物细胞充分分散,从土壤中分离出来。此为10-1的土壤悬液,吸取1 mL此土壤悬液于9 mL无菌水中,另用无菌吸管吹吸3次混匀,制成10-2土壤悬液。以此类推,制成10-3、10-4、10-5、10-6、10-7、10-8不同稀释度的土壤悬液。然后根据菌群选择合适的稀释度来进行实验,本文采用的是稀释平板法,分别设置三个浓度梯度,三次重复。最后用平板接种法将菌种接至平板培养基上(用移液管吸取一定体积的菌液移至平板培养基上)进行培养。其计数方法为涂布平板计数法,设置三个重复,分别编号,把配好的培养基分别倒入培养皿中,凝固,然后移液枪吸取不同稀释度的悬液放在编好号的培养基中间。用无菌玻璃棒均匀的在平板表面上轻轻的涂布。培养条件是把在无菌操作台上涂布好的平板再次用紫外线杀菌,然后放入恒温冰箱(25℃-28℃)进行培养,至菌落长出后即可计数。培养时间由短到长分别是:细菌1~2 d,真菌3~4 d,放线菌5~7 d。细菌的培养—牛肉膏蛋白胨琼脂培养基;真菌的培养—马丁-孟加拉红琼脂培养基;放线菌的培养—改良高氏1号琼脂培养基。
微生物计算公式:cfug-1=MD/W
式中M为菌落平均数;D为稀释倍数;W为土壤烘干质量
土壤酶的测定方法:用苯酚钠和次氯酸钠对脲酶进行比色,过氧化氢酶活性采用高锰酸钾滴定法,蔗糖酶活性采用3,5-二硝基水杨酸比色法测定,酸性磷酸酶活性采用硝基酚比色法。
123数据处理
实验结果均用3次重复所得数据的平均值进行分析。用Excel 2010进行数据的统计、初步处理和作图,用SPSS 170软件分别对微生物和土壤酶进行显著性分析。
2结果与分析
21不同间作模式下对刺梨园土壤微生物类群的数量特征
211不同间作模式下土壤细菌的数量从图1中可以看出,两种间作模式下土壤细菌数量高于刺梨单作,即间作对土壤细菌的生长有促进作用。比较结果为:辣椒//刺梨>玉米//刺梨>刺梨。在土壤深度0~20 cm,BO和AO的土壤细菌数量显著高于CK(P<005),其中,AO是CK的364倍,BO是CK的294倍;AO和BO间没有显著差异(P>005),但AO较BO有上升的趋势,其增加的百分数为2371%;在土壤深度20~40 cm,AO和BO的细菌含量显著高于CK(P<005),其中,AO是CK的251倍,BO是CK的219倍; BO和AO的细菌数量没有显著差异性(P>005)。如图所示,随着土层深度的增加,细菌数量是逐渐降低的。
212不同间作模式下土壤真菌的数量
如图2所示,间作模式下土壤真菌含量均高于单作,即间作对土壤真菌生长有促进作用。比较结果为:玉米//刺梨>辣椒//刺梨>刺梨。在土壤深度0~20 cm,AO和BO与CK之间土壤真菌数量有着显著差异性(P<005),其中,AO是CK的165倍,BO是CK的183倍;BO和AO之间有显著差异(P>005)。在土壤深度20~40 cm,AO和BO的土壤真菌含量显著高于CK(P<005),AO是CK的185倍,BO是CK的2倍。其中,BO较AO有上升趋势,增加的百分数为833%;从上图可知,土壤真菌的数量随着土层深度的增加呈逐渐下降趋势,0~20 cm层的真菌数量较多。
213不同间作模式下土壤放线菌的数量
如图3可知,间作模式下土壤放线菌的含量高于刺梨单作,即间作对土壤放线菌的生长有促进作用。比较结果为:玉米//刺梨>辣椒//刺梨>刺梨。在土壤深度0~20 cm,AO和BO与CK之间土壤放线菌的含量有着显著差异性(P<005),其中,AO是CK的140倍,BO是CK的162倍;BO较AO的含量有增长趋势,其增加的百分数为1593%。在土壤深度20~40 cm,AO和BO的土壤放线菌的含量显著高于CK(P<005),其中,AO是CK的133倍,BO是CK的138倍,BO和AO的放线菌含量没有显著差异性(P>005),有增长的趋势,增长的百分数为375%。由图中还可以看出来,土壤放线菌的数量随着土层深度的增加而逐渐减小。
LEfSe组间群落差异分析。即linear discriminant analysis(LDA),线性判别分析。LEfSe根据分类学组成对样品按照不同的分组条件进行线性判别分析,找出对样品划分产生显著性差异影响的群落或物种。
2结果与分析
21肠道微生物Beta多样性分析
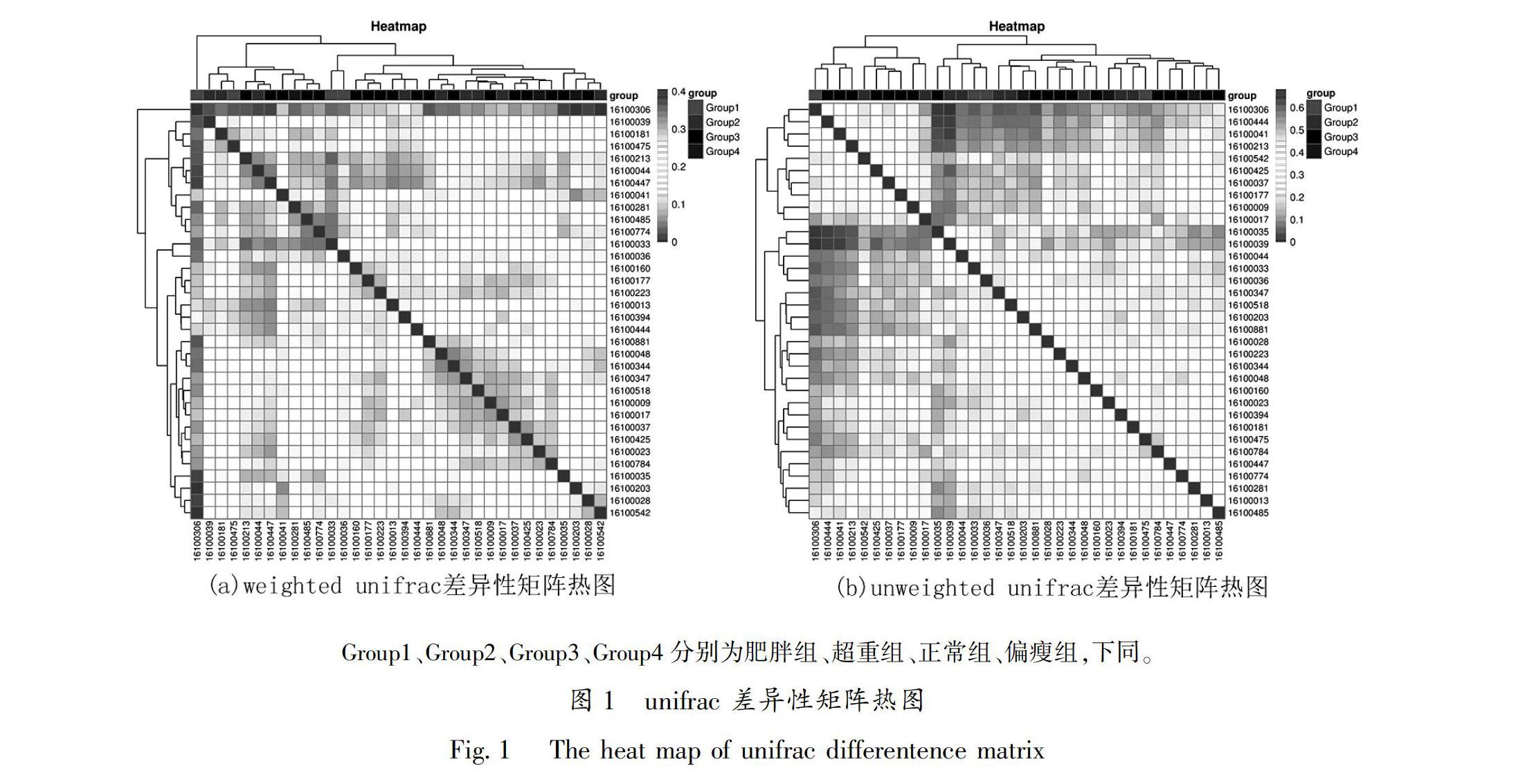
211UniFrac分析
对样品两两之间进行比较分析,得到样品间的UniFrac距离矩阵,进行可视化操作,能够得到UniFrac差异性矩阵热图(见图1)。图1可以看出,肥胖与偏瘦组差异性较强,肥胖组与超重组、超重组与正常组间差异较小。
212基于距离矩阵构建的PCoA
由图2可观察到,肥胖组与正常组、肥胖组与偏瘦组差异较大,正常组与超重组有部分重合,菌群组成有一定相似性,正常组与偏瘦组重合较多,相似性较强。超重组与肥胖组在图2b中有部分重合,显示具有一定相似性。
213多样品相似度树状图
由图3可观察到,不同样品中,肥胖组、超重组、正常组各组间的肠道微生物菌群在进化上存在差异性,但肥胖组与超重组、正常组与肥胖组也存在一定的相似性。
214基于UniFrac的NMDS非度量多维尺度分析
从图4a基于Weighted UniFrac NMDS看,肥胖组与偏瘦组距离较远,散布性强,差异性较大,肥胖与正常组重合度较高,差异较小;而图4b Unweighted UniFrac NMDS 难以明显区分。
215基于物种信息的Bray ̄Curtis距离分析
在门水平,肥胖组与其他组距离较远,正常组与超重组距离较近;在种属水平上,肥胖组与超重组距离较近,差异较小(见图5)。
22肠道微生物构成与丰度变化
221群落物种分布柱状图
在属水平上,鉴定出99个属(见图6a),其中相对丰度大于100%的属包括:拟杆菌属(Bacteroides 4168%,Faecalibacterium 646%)、考拉杆菌属(Phascolarctobacterium 176%)、巨单胞菌属(Megamonas 166%,Parabacteroides 273%,Parasutterella190%,梭菌属(Fusobacterium 212%)、毛螺菌属(Lachnospira136%)、Alistipes(118%)、萨特菌属(Sutterella 103%,Subdoligranulum 133%)和羅氏菌属(Roseburia 158%)。共占测序序列总数的6478%。
在种水平上(见图6b),相对丰度大于100%的属包括:单形拟杆菌(Bacteroides uniformis 344%,Bacteroides massiliensis 238%)、脆弱拟杆菌(Bacteroides fragilis 383%,Fusobacterium mortiferum188%)。
222热图(见图7)
从图7中可以看出,门水平,Actinobacteria、Fusobacteria、Proteobacteria颜色差异明显;纲水平,Deltaproteobacteria、Coriobacteriia、Betaproteobacteria、Bacilli、Erysipelotrichia颜色变化较大;目水平,Fusobacteriales、Pasteurellales、Coriobacteriales、Burkholderiales、Lactobacillales、Erysipelotrichales、Enterobacteriales颜色变化较大;科水平Prevotellaceae、Alcaligenaceae、Acidaminococcaceae、Pasteurellaceae、Rikenellaceae、Streptococcaceae、Desulfovibrionaceae、Peptostreptococcaceae、Veillonellaceae、Coriobacteriaceae、Erysipelotrichaceae颜色变化较大;属水平Erysipelatoclostridium、Romboutsia、Megamonas、Parasutterella、Phascolarctobacterium、Anaerostipes、Subdoligranulum、Flavonifractor、Streptococcus、Lachnospira、Dorea 颜色范围较广。
23LDA EffectSize 组间群落差异分析
图8a显示,正常组中,Cyanobacteria、Betaproteobacteria起到重要作用;肥胖组中,Erysipelotrichaceae、Erysipelotrichales、Erysipelotrichia起到重要作用;超重组中,Pseudomonadales起到重要作用。
从图9可以观察到,o_Bifidobacteriales与g_Bifidobacterium,在正常组中丰度最高,肥胖组与超重组丰度较低,在偏瘦组中未检测到;g_Parabacteroides在超重组丰度最高,正常组、肥胖组、偏瘦组均有检测到;p_Cyanobacteria在正常组丰度最高,肥胖组与超重组丰度较低,在偏瘦组中未检测到;s_uncultured_Lachnospiraceae_bacterium在偏瘦组中丰度最高,肥胖组与超重组丰度较低,正常组中个别样本丰度极高,整体丰度较低;g_Terrisporobacter在肥胖组丰度极高,超重组丰度较高,在正常组、偏瘦组丰度极低;c_Erysipelotrichia与o_Erysipelotrichales在肥胖组丰度较高,超重组、正常组、偏瘦组丰度较低;f_Erysipelotrichaceae在肥胖组丰度最高,其他组中丰度较低;g_Candidatus_Stoquefichus在偏瘦组丰度较高,在肥胖组、超重组、正常组丰度较低;g_Holdemanella在肥胖组丰度较高,正常组丰度较低,超重组与偏瘦组丰度极低;g_Megamonas在肥胖组丰度较高,超重组丰度较低,正常组与偏瘦组丰度极低;s_uncultured_bacterium在肥胖组最高,超重组、正常组与偏瘦组丰度较低;c_Betaproteobacteria在正常组最高,超重组与偏瘦组丰度较低,肥胖组丰度最低;g_Bilophila和s_Bilophila_wadsworthia在偏瘦组丰度最高,正常组丰度较低,肥胖组与超重组丰度极低;o_Pseudomonadales在超重组丰度最高,肥胖组与正常组丰度较低,偏瘦组丰度极低。
3结论与讨论
在菌群构成上,丰度最高的是拟杆菌门(Bacteroidetes),其次為厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、梭杆菌门(Fusobacteria),放线菌门(Actinobacteria),这5个菌门在肠道微生物群中占据绝对优势。
随着BMI值的升高,肠道微生物群的多样性和丰度呈现出差异。丰度数据显示,肥胖组与其他三组,特别是偏瘦组的差异巨大。对比超重组和正常组发现,二者具有显著丰度差异的菌群,大多数是超重组低于正常组。在菌群构成多样性上,肥胖组中菌群多样性较低,超重组和正常组差异较小。肠道微生物是一个动态平衡的系统,系统内菌群间的关联较为复杂,具有一定的自我恢复能力,因此,超重组人群肠道微生物构成的改变可能慢于BMI的提升,更倾向于正常组的肠道微生物构成,或者在此阶段,肠道微生物的结构变化对肥胖的发生影响较小,饮食、运动等因素对BMI的影响较大。
LEfSe分析显示:肥胖组中,Erysipelotrichaceae、Erysipelotrichales、Erysipelotrichia起着重要作用;正常组中,Bifidobacteriales、Cyanobacteria、Betaproteob ̄acteria起着重要作用;超重组中,Pseudomonadales起着重要。结合丰度差异,显示肥胖与肠道微生物中厚壁菌丰度变化的关联更多在科属水平,肥胖组种起着重要作用的细菌属于厚壁菌门,而门水平上拟杆菌门丰度变化显著,可能厚壁菌门与拟杆菌门共同作用对肥胖的影响更大;正常组中,双歧杆菌发挥重要作用,维护肠道。
参考文献:
[1]TURNBAUGH P J, LEY R E, HAMADY M, et al. The Human Microbiome Project [J]. Nature, 2007, 449 (7164) : 804-810.
[2]ABUBUCKER S, SEGATA N, GOLL J, et al. Metabolic Reconstruction for Metagenomic Data and Its Application to the Human Microbiome[J]. PLoS computational biology, 2012, 8(6):e1002358.
[3]HUTTENHOWER C, GEVERS D, KNIGHT R, et al. Structure. function and diversity of the healthy human microbiome[J]. Nature, 2012, 486(7402):207-214.
[4]CONSORTIUM H M P. A framework for human microbiome research [J]. Nature, 2012, 486(7402):215-221.
[5]PEEK R M, BLASER M J. Helicobacter pylori and gastrointestinal tract adenocarcinomas[J]. Nature reviews cancer, 2002, 2(1):28-37.
[6]LEY R E, BACKHED F, TURNBAUGH P, et al. Obesity alters gut microbial ecology[J]. Proceedings of the national academy of sciences of the united states of america, 2005, 102(31):11070-11075.
[7]TURNBAUGH P J, LEY R E, MAHOWALD M A, et al. An obesity ̄associated gut microbiome with increased capacity for energy harvest[J]. Nature, 2006, 444(7122):1027-1031.
[8]BLASER M J, Chen Y, REIBMAN J. Does Helicobacter pylori protect against asthma and allergy [J]. Gut,2008, 57(5):561-567.
[9]ZHAN G, CHI ̄HONG T, STROBER B E, et al. Substantial Alterations of the Cutaneous Bacterial Biota in Psoriatic Lesions[J]. PLoS one, 2008, 3(7):e2719.
[10]ISLAMI F, KAMANGAR F. Helicobacter pylori and esophageal cancer risk: a meta ̄analysis. Cancer Prev[J]. Res, 2008, 1(5):329-338.
[11]GARRETT W S, GALLINI C A, YATSUNENKO T, et al. Enterobacteriaceae Act in Concert with the Gut Microbiota to Induce Spontaneous and Maternally Transmitted Colitis[J]. Cell host & microbe, 2010, 8(3):292-300.
