基于Stacking算法与多数据源的有效药物组合
2020-05-25朱永先
朱永先
摘 要:对于癌症、心血管疾病等复杂疾病,采取组合用药克服耐药性和改善功效已成为标准治疗方案。鉴定药物组合标准的方法是进行体内或体外药物筛选实验,但这一过程很缓慢,代价高昂。各种高通量组学技术产生度量药物效应的各层次数据,使得从计算角度挖掘数据进而预测有效药物组合成为主流手段。针对有效药物组合的预测模型大多是利用单一机器学习模型建模。为获得更高的精度,提出一种新的有效药物组合预测方法。该方法充分利用5种不同层次的药物信息构建相似性特征,特别引入药物靶标的序列信息和功能信息,基于Stacking算法融合多个传统机器学习模型和最新的集成学习模型LightGBM。实验表明,该方法预测的AUC值为0.953,精度比单一机器学习模型有显著提升。
关键词:药物组合;模型融合;基分类器;预测方法;相似性特征
DOI:10. 11907/rjdk. 191508 开放科学(资源服务)标识码(OSID):
中图分类号:TP306文献标识码:A 文章编号:1672-7800(2020)002-0100-05
英标:Effective Drug Combination Based on Stacking Algorithm and Multi-data Source
英作:ZHU Yong-xian
英单:(Business School, University of Shanghai for Science and Technology, Shanghai 200093, China)
Abstract: For complex diseases such as cancer and cardiovascular diseases, it has become a standard treatment to use combination drugs to overcome drug resistance and improve efficacy. The method for identifying drug combination criteria is through in vivo or in vitro drug screening experiments, but this process is expensive and slow. Various high-throughput omics techniques produce data at various levels that measure drug effects, making it a mainstream and effective means of mining data from a computational perspective to predict effective drug combinations. Most of the current predictive models for effective drug combinations are modeled using a single machine learning model. In order to obtain a better prediction rate, this paper proposes a new effective drug combination prediction method. The method makes full use of five different levels of drug information to construct similarity features, especially the introduction of sequence information and functional information of drug targets. Based on the Stacking algorithm, multiple traditional machine learning models and the latest integrated learning model LightGBM are combined. Finally, the AUC value of the prediction result of this method is as high as 0.953, and the precision is significantly improved compared with the single machine learning model.
Key Words: drug combination; model fusion; base classifier; prediction method; similarity feature
0 引言
组合用药指两种或多种有效药物成分组合在一起,共同治疗某种疾病。在治疗癌症和心血管疾病时,组合用药比单药治疗效果更好。药物组合的多种成分会同时作用多种不同靶蛋白,能更好地避免复杂疾病所涉及的反馈机制,即提高复杂疾病的治疗效果并降低副作用,这是一种非常有前景的治疗策略[1-5]。然而,考虑药物之间所有的可能匹配,经实验筛选新的药物组合是不切实际的[6]。随着各种组学技术的发展,积累了大量生物学数据,运用生物信息学和计算工具,挖掘并整合这些数据中相互关联信息,从中筛选出有效的药物组合,既符合成本效益,又节约人力时间[7]。当然,现有大多数方法都是为了预测两种药物的协同效应,因为三种药物组合效应在技术上难以预测,缺乏模型评价的实验数据。
有效药物组合预测研究近年取得了不俗成果。模型的特征数据分为静态数据和动态数据。静态数据包括药物的生物学信息(靶蛋白,pathway)、化学信息、药理学信息;动态数据包括基因表达数据、蛋白质相互作用数据(PPI)。Sun等[8]運用基因表达数据构建生物学特征,利用支持向量机及朴素贝叶斯分类器预测有效药物组合;Li等[9]基于生物学信息和基因表达数据构建生物分子网络,利用随机森林算法预测有效药物组合;Preuer[10]等使用化学信息和基因表达数据作为输入,利用深度学习算法预测有效药物组合。Xu等[11]整合生物学、化学及药理学信息,运用随机梯度提升算法预测药物组合。
结合药物的动静数据构建特征,并利用机器学习算法进行有效组合预测效果突出,但这些预测方法大多使用单一的机器学习模型。为了增强模型的鲁棒性,提升预测精度,本文应用Stacking算法融合主流且强大的机器学习算法构建药物组合预测模型。对于特征的构建,除利用常用药物动静态信息外,还深入挖掘药物靶标信息,引入靶标的序列信息和功能(Go Term)信息。
1 Stacking模型融合算法
1.1 Stacking简介
Stacking(堆叠)是Wolpert[12]于1992年在“Staked Generalization”论文中提出的一种集成学习算法。不同于bagging和boosting采用相同的分类算法训练单个学习器[13-14],Stacking能够组合来自多个预测基模型的结果信息生成新模型,是一种特殊的组合策略。通常Stacking模型融合可以突出效果好的基模型,同时抹黑执行效果不佳的基模型,所以预测效果优于单个模型。特别当基模型显著不同时,堆叠是最有效的。
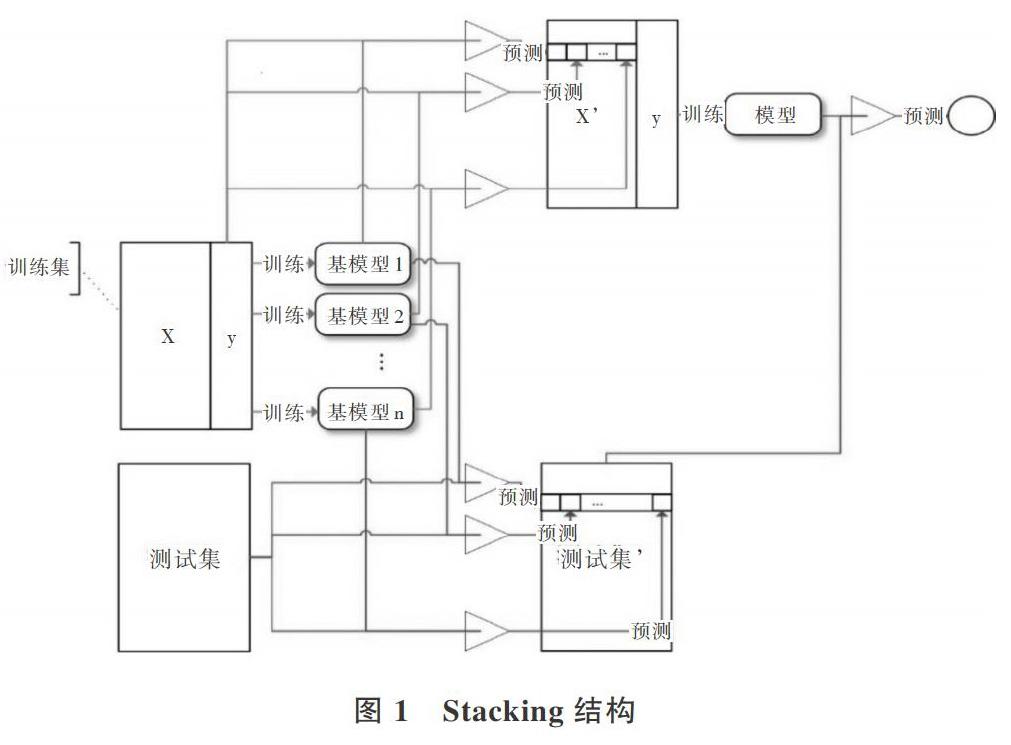
Stacking将基模型的输出结果作为新特征输入到其它模型中,这种方法实现了模型的堆叠,即以第一层的模型输出作为第二层模型的特征输入,第二层模型的输出作为第三层模型的特征输入,依此类推,最后一层模型输出的结果作为最终预测结果,最后训练出精准、稳健、鲁棒的模型。Stacking一般为两层结构,如图1所示。
1.2 Stacking选择
本文综合应用药物生物学、化学、药理学等多方面信息构建药物组合特征,提升有效药物组合的预测精度。运用Stacking算法融合主流且具有显著差异的机器学习算法,通过组合几种机器学习技术组成一个预测模型,以达到减小方差、偏差和提升预测精度的效果。研究选用逻辑回归(LR)、随机森林(Random Frost)、K-近邻算法(KNN)和高效梯度提升决策树(LightGBM)4个机器学习算法进行Stacking融合。
1.3 基分类器
Stacking模型融合需要多个基分类器构成差异化,根据Stacking的特性本文选用以下4个基分类器。
1.3.1 随机森林
随机森林算法(Random Frost)是基于Bagging 集成学习理论的代表算法,由Leo Breiman[15]于2001提出。主要思想是从给定的数据集 K 中随机抽取 m 个样本用以生成新的训练样本集合,然后根据新的训练样本集生成 m 个决策树,同时这m个决策树为了避免过拟合,不进行后剪枝处理,单个分类结果按各个决策树投票多少形成的分数决定。一棵决策树的分类能力可能很小,但在随机产生大量决策树并组成随机森林后,m个样品都逐一通过一棵树分类决策,最后组合的结果将更接近于正确分类。随机森林通过在每个节点处随机选择特征进行分支,每棵分类树之间的相关性得到最小化,故对多元共线性不敏感,这样就提高了分类的精确性及抗噪声能力。
1.3.2 K-近邻算法
K-近邻算法(KNN)是基于实例的代表算法,1968年由Cover & Hart[16]提出,是一种用于分类和回归的无母数统计方法,主要思想是通过测量不同特征值之间的距离进行分类。如果一个样本在特征空间中的k个最相似(特征空间中最邻近)的样本中的大多数属于某一个类别,则该样本也属于这个类别。KNN 方法虽然依赖于极限定理,但在类别决策时只与极少量的相邻样本有关。KNN算法中,所选择的邻居都是已经正确分类的对象。该方法在定类决策上只依据最邻近的一个或几个样本的类别决定待分样本所属的类别。
1.3.3 邏辑回归
逻辑回归(LR)是回归算法的代表,由统计学家David Cox[17]于1958年开发。它是广义线性模型的特殊应用,是一个二分类问题预测方法。LR使用 Sigmod 函数作为预测函数,估计预测结果概率 P(y | x)的大小。如果 P > 0.5则认为属于正类别,否则属于负类别。在有效药物组合预测问题中,Sigmod函数的输出就是每对药物组合是否有效的概率值,大小取值在 0-1 之间。Logistic 模型在训练阶段,通过随机梯度下降法(SGD)不断最小化预测函数误差以提高模型的泛化能力。为避免模型陷入过拟合,在损失函数上采用相应的正则化方式,以缓解模型的过拟合程度。
1.3.4 高效梯度提升决策树
高效梯度提升决策树(LightGBM)是基于GBDT 树和Boosting 算法的分布式机器学习框架。LightGBM 是微软DMTK 团队2016年的开源项目,为Gradient Boosting(GBDT) 算法的改进版本[18]。其中 GBDT 算法的思想是将弱分类算法提升为强分类算法,从而在一定程度上提高分类准确率。LightGBM中的决策树子模型采用按叶子分裂的方法分裂节点,因此它的计算代价较小。也正是因为选择了这种分裂方式,需要控制树的深度和每个叶子节点的最小数据量,从而避免过拟合现象发生。LightGBM 选择基于Histogram的决策树算法,将特征值分为很多个小 “桶”,进而在这些 “桶”上寻找分裂,这样可以降低储存成本和计算成本。LightGBM与目前最为强大的机器学习模型XGBoost相比,速度更快、内存占用更少、准确率更高,所以本研究选取其作为最后一层的分类器。
2 特征工程
2.1 数据准备
本文有效药物组合数据来自Drug Combination Database(DCDB,version 2.0)[19](http://www.cls.zju.edu.cn/dcdb/),最新版本的DCDB收集了1 363种药物组合(330份批准和1 033份研究,包括237份无效用法),涉及904种个体药物,805种靶标。将946种成对药物组合作为研究对象,通过爬虫技术爬取DrugBank数据库得到单药的二维分子结构信息、靶标信息、ATC编码信息[20],其中各个靶标的序列信息和Go Term信息分别下载自Uniprot与STRING数据库。如果一种药物以上信息不全,其涉及的药物组合将被删除。通过筛选,共留下358对药物组合。根据DCDB提供的药物组合清单信息,358对药物组合有效的有317对,无效的41对,故获得标准正样本317份,标准负样本41份。
2.2 特征构建
通过整合药物的二维分子结构信息、药物ATC编码信息、蛋白质相互作用、药物靶标序列信息和Go Term信息,构建5个不同层次的药物组合特征。
2.2.1 二维分子结构相似性特征
本文应用软件RDKit计算两个药物小分子之间的相似度。 RDKit是一款免费开源的化学信息学与机器学习软件,提供了C++和Python的API,利用Python脚本很容易计算多个分子或构象之间的接近程度。首先利用获取的各个药物分子二维结构信息(SMILES),调用RDKit计算出对应的二维 MACCS 分子指纹[21],然后用谷本系数(Tanimoto)计算两种药物二维 MACCS 分子指纹相似度。药物[Di]和[Dj]的Tanimoto系数定义如下:
式(1)中,[Mi]和[Mj]分别代表药物[Di]、[Dj]的二维 MACCS 分子指纹。
2.2.2 ATC编码相似性特征
ATC编码是解剖治疗学及化学分类系统编码,可以代表药物的疗效信息。ATC编码共有7位,其中第1、4、5位为字母,第2、3、6、7位为数字。 ATC系统将药物分为5个级别:ATC编码第1级为一位字母,表示解剖学上的分類,共有14个组别;ATC编码第2级为两位数字,表示治疗学上的分类;ATC编码第3级为一位字母,表示药理学上的分类;ATC编码第4级为一位字母,表示化学上的分类;ATC编码第5级为两位数字,表示化合物上的分类。两种药物成分的第 K 级药物疗效相似性定义如下:
在此, ATC 编码在第 N 级水平上表示为[ATCN(Di)],极少的药会在ATC 编码的5级水平上都一致,故N取3级。本文的ATC编码相似性特征为两种药物ATC编码相似度的最大值。
2.2.3 蛋白质相互作用的相似性特征
本文从 STRING 数据库(Version 11.0)(https://string-db.org/)下载所有人类两两靶标的蛋白质相互作用得分[22],将成对药物对应的靶标与之匹配,并累加两药靶标间所有蛋白质相互作用得分,最后取平均分值,得到药物组合蛋白质相互作用相似性得分,定义如下:
式(4)中,[pi]和[pj]分别代表药物[Di]、[Dj]的靶标。
2.2.4 药物靶标序列相似度特征
利用Biopython(http://www.biopython.org)进行药物靶标的序列相似度计算。Biopython是为生物信息学开发者提供的在线资源库,包括模块、脚本以及一些基于Python的软件网站链接。Biopython提供了独立的模块pairwise2进行成对序列比对。同样,药物靶标的序列相似度得分与蛋白质相互作用相似性得分类似,如式(4)所示。
2.2.5 药物靶标的Go Term相似性特征
使用软件EnrichFunSim[23]构建两两药物靶标的Go Term相似性特征。基因之间的功能相似性广泛应用于生物信息学,评估基因功能相似性的方法主要是基于基因本体术语(Go Term)的语义相似性。EnrichFunSim是一种将Go结合现有功能的最新方法。实验表明,EnrichFunSim显著提高了功能相似性测量能力。
3 实验与分析
3.1 实验数据处理
本文构建的特征数据都是数值类型,其大小有实际意义,表示相似度高低。因为不同特征数值量级不同,因此需要进行标准化处理消除特征之间的数量级差别,以避免数值问题,平衡各个特征贡献,提升模型求解速度。标准化公式如下:
考虑到总样本358份,其中标准正样本317份,标准负样本41份,预测成对药物组合是否有效成为一个二分类问题。为克服正负样本不平衡问题,本研究采用SMOTE算法进行过采样。SMOTE算法的思想是合成新的少数类样本,合成方式是对每个少数类样本a从它的最近邻中随机选一个样本b,然后在a、b之间的连线上随机选一点作为新合成的少数类样本。
3.2 实验分析
3.2.1 基模型建模分析
首先利用构建的特征数据和全部正负样本,将总数据中67%划分为训练集,其余作为测试集。分别输入逻辑回归(LR)、随机森林(Random Frost)、K-近邻算法(KNN)和高效梯度提升决策树(LightGBM)4个模型中,结果如表1和图2所示。
3.2.2 Stacking模型融合分析
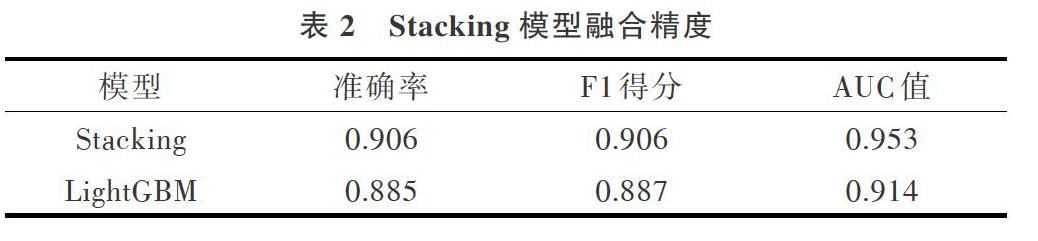
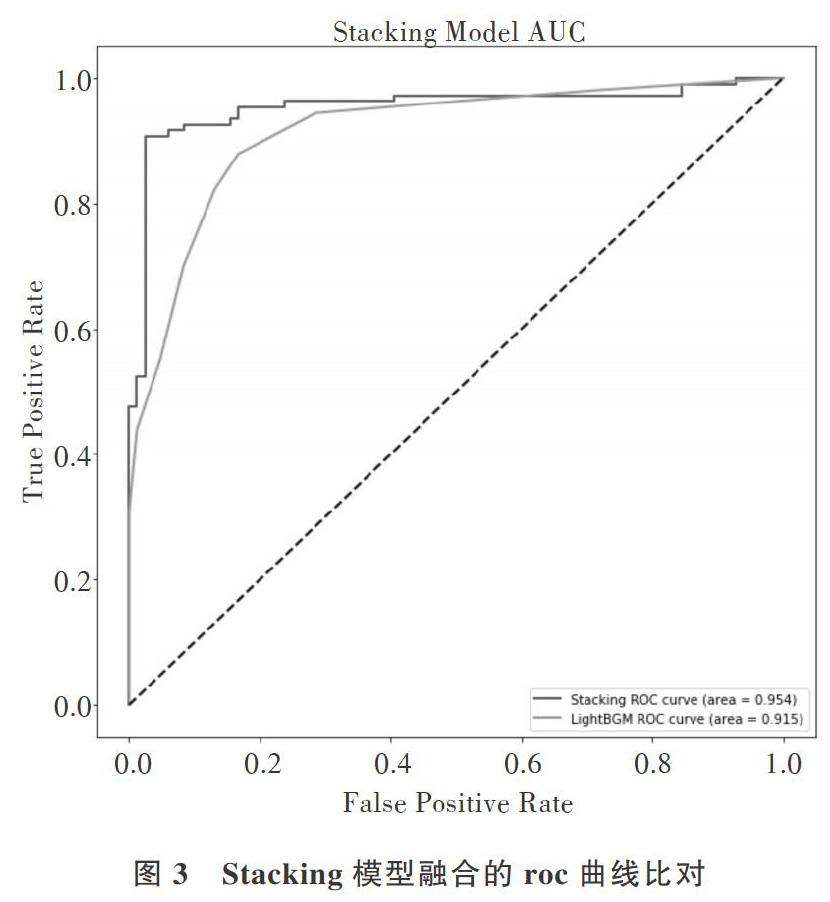
Stacking模型融合过程:第一层在LR、Random Frost、 KNN三个基分类器上,分别对训练数据作5折交叉验证。对第一个基分类器,先从训练集拿出4折作为训练数据,另外1折作验证数据,用4折数据训练好的模型去预测另外1折验证数据,得到概率结果为[P1_n(n=5)]。同时测试数据没有加入5折交叉验证,所以每次用此模型去预测对应测试数据,得到[T1_n(n=5)],最后测试数据输出的[Tn]取平均值为[T1_MEAN],拼接[P1_n]与[T1_MEAN]为新的特征数据[[P1_1,P1_2,P1_3,P1_4,P1_5,T1_MEAN]]。接下来对其它两个基分类器采用同样的策略产生新的特征数据[[P2_1,P2_2,][P2_3,P2_4,P2_5,T2_MEAN]]和[[P3_1,P3_2,P3_3,P3_4,P3_5,][T3_MEAN]]。第二层,利用前面3个基分类器产生的新的特征数据输入到LightGBM分类模型中,模型融合结果如表2、表3和图3所示。
由于单模型LightGBM预测效果最好,故取其与Stacking模型融合进行对比分析。由表2和图3可以发现,基于Stacking模型融合的药物组合预测模型预测精度明显优于基于LightGBM的药物组合预测模型,部分预测结果见表3。
4 结语
本研究综合应用了药物二维分子结构信息、药物ATC编码信息、蛋白质相互作用、药物靶标的序列信息和Go Term信息,对药物信息的提取与集成突出了层次性、互补性。同时引入Stacking算法,融合逻辑回归(LR)、随机森林(Random Frost)、K-近邻算法(KNN)和高效梯度提升决策树(LightGBM)4个机器学习算法构建药物组合预测模型。实验结果表明,利用Stacking算法模型融合的预测效果优于传统及最新的机器学习算法建模。但本文还有一些局限性,如利用SMOTE算法进行过采样解决正负样本不平衡问题,由此生成的并不是真正的负样本,存在生成的负样本为有效药物组合的情况,如何寻找更多真正的负样本或者考虑不利用负样本建模是未来的研究方向。
参考文献:
[1] SUCHER NIKOLAUS J. Searching for synergy in silico, in vitro and in vivo[J]. Synergy, 2014,1(1):30-43.
[2] CHOU T C . Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies[J]. Pharmacological reviews, 2006,58(3):621-681.
[3] SHIM JOONG SUP, LIU JUN O. Recent advances in drug repositioning for the discovery of new anticancer drugs[J]. Int J Biol Sci, 2014, 10(7):654-663.
[4] GU L, LIU H, FAN L,et al. Treatment outcomes of transcatheter arterial chemoembolization combined with local ablative therapy versus monotherapy in hepatocellular carcinoma: a meta-analysis[J]. Cancer Res Clin Oncol. 2013(140):199–210.
[5] ZHANG X, ZHANG X J, ZHANG T Y, et al. Effect and safety of dual anti-human epidermal growth factor receptor 2 therapy compared to monotherapy in patients with human epidermal growth factor receptor 2-positive breast cancer: a systematic review[J]. BMC Cancer,2014(14):625-630.
[6] DIMASI JOSEPH A, GRABOWSKI HENRY G, HANSEN RONALD W. Innovation in the pharmaceutical industry: new estimates of R&D costs[J]. Journal of health economics, 2016(47):20-33.
[7] 趙明珠. 药物—靶标相互作用及药物对组合研究[D]. 上海:上海交通大学,2013.
[8] SUN Y, XIONG Y, XU Q, et al. A hadoop-based method to predict potential effective drug combination[J]. BioMed research international, 2014(6):1541-1548.
[9] LI X Y,QIN G R,YANG Q M,et al. Biomolecular network-based synergistic drug combination discovery[J]. Biomed Research International, 2016 (2):1-11.
[10] KRISTINA PREUER,RICHARD P I LEWIS. Deepsynergy: predicting anti-cancer drug synergy with Deep Learning[J].Bioinformatics, 2017(11):1-9.
[11] XU Q, YI X, OU H Y,et al. A similarity-feature based method to predict effective drug combination[J]. Interdisciplinary Sciences Computational Life Sciences,2015(3):896-907.
[12] WOLPERTDH. Stacked generalization[J]. NeuralNetworks,1992,5(2):241-259
[13] BREIMANL. Baggingpredictors[J]. MachineLearning,1996,24(2):123-140.
[14] BREIMANL. Arcingclassifiers[J]. AnnalsofStatistics,1998,26(3):801-849
