加压流体萃取-气质联用法测定土壤中15种多环芳烃
2020-03-24林晶
林晶

摘 要:建立加压流体萃取-气质联用法用于测定土壤中15种多环芳烃,对萃取溶剂、净化洗脱、分析温度等条件进行优化比选,采用不分流和选择离子模式进行测定。结果表明,此方法在5~1000μg/L范围内,15种多环芳烃相对响应因子的相对标准偏差均小于20%;方法检出限为0.1~0.4μg/kg,回收率为41.7~128%,相对标准偏差2.6%~13.3%。方法灵敏度高,优于现有标准,准确度和精密度均满足多环芳烃批量痕量检测要求。
关键词:土壤;多环芳烃;气质联用法;优化
中图分类号:X833 文献标志码:A 文章编号:2095-2945(2020)09-0121-05
Abstract: A pressurized fluid extraction-GC-MS method was established for the determination of 15 kinds of polycyclic aromatic hydrocarbons in soil. The extraction solvent, purification and elution and analysis temperature were optimized and determined by non-shunt and selective ion mode. The results show that in the range of 5~1000 μg/L, the relative standard deviations of the relative response factors of 15 polycyclic aromatic hydrocarbons are all less than 20%; the detection limit of the method is 0.1~0.4 μg/kg, the recovery is 41.7% 128%, and the relative standard deviation is 2.6%~13.3%. The method has high sensitivity and is superior to the existing standards, and its accuracy and precision can meet the requirements of batch trace detection of polycyclic aromatic hydrocarbons.
Keywords: soil; polycyclic aromatic hydrocarbons; GC-MS (Gas Chromatograph-Mass Spectrometer); optimization
多环芳烃(Polycyclic Aromatic Hydrocarbons,PAHs)是煤、石油、木材、烟草、有机高分子化合物等有机物不完全燃烧时产生的含多个苯环结构的碳氢化合物[1-2],因其致癌、致畸、致突变的毒理特性,已越来越多地受到国内外环保专家学者的关注。土壤作为生态系统中的基底载体,容易将自然环境中以气态或被大气粉尘吸附等方式存在的PAHs通过干湿沉降方式带入土中[3],PAHs的脂溶性特点使其容易进入生物体内,经过生物链的迁移、转化、富集,最终对人类健康和安全造成威胁[4]。
目前多环芳烃检测方法主要有高效液相色谱法和气相色譜质谱法[5-6],其中气相色谱质谱法利用气相色谱仪的高分离特性和质谱仪对单一组分的准确鉴定特性,能够快速有效检测出多组分多环芳烃。前处理方法有索氏提取法、超声萃取法、微波萃取法、加压流体萃取法等[7-8],其中加压流体萃取法是目前较为先进的处理方法,因其溶剂使用量相对少、提取速度较快等优点而被广泛使用。故本文选取加压流体萃取-气质联用法为检测方法。但土壤实际样品分析中还存在不少问题,由于土壤基质成分复杂,分析干扰大,且土壤中的PAHs往往是痕量存在[9],不少方法灵敏度无法满足实际工作需要,例如现有的HJ805-2016《土壤和沉积物多环芳烃的测定气相色谱质谱法》中就是SCAN检出限,检出限值较大,无法满足普通土壤样品的微量检测要求;同时实验过程中大剂量有机溶剂的使用,很容易对环境造成二次污染。因此实验过程中不断总结经验,优化各个环节至关重要。本文以提高检测灵敏度、降低方法检出限,以及获得更优的性能条件、更小的环境污染为目标,探索实验中的提取、净化及仪器分析优化手段,实现对土壤中苊烯、苊、芴、菲、蒽、荧蒽、芘、苯并[a]蒽、 、苯并[b]荧蒽、苯并[k]荧蒽、苯并[a]芘、茚并[1,2,3-c,d]芘、二苯并[a,h]蒽和苯并[g,h,i]苝等15种多环芳烃的精准微量测定。
1 实验部分
1.1 仪器与试剂
气相质谱联用仪(安捷伦7890A-5975C型)、加速溶剂萃取仪(Dionex ASE350)、氮吹浓缩仪(Biotage TurboVapⅡ)、真空冷冻干燥仪(空载真空度8Pa,北京博医康实验仪器有限公司)、固相萃取仪(SUPELCO-6位萃取装置)、高纯氦(φ(He)=99.999%)、高纯氮(φ(N2)=99.999%)、安捷伦HP-5MS色谱柱(长30m,内径0.25mm,膜厚0.25μm)。
正己烷(色谱纯,SupraSolv)、二氯甲烷(色谱纯,OCEANPAK)、丙酮(色谱纯,OCEANPAK)、石英砂(150μm(100目),科密欧公司)、无水硫酸钠(优级纯,科密欧公司)、硅酸镁净化小柱(1000mg,柱体积6mL,Agela Technologies)、内标物为苊-d10、菲-d10、 -d12和苝-d12(Accu Standard,Inc Z-014J-0.5X-1ml(217031403)(ρ=2000mg/L))、替代物为2-氟联苯和对三联苯-d14(o2si 114006-01-1mL(308102)(ρ=2000ug/mL))、多环芳烃标准溶液(Accu Standard,Inc Z-0.13-17-1ml(217041359)(ρ=200mg/L))。
1.2 样品制备
采集土壤样品,放置于搪瓷盘中,将其中的碎石、枝叶等异物去除,研磨混匀后放入真空冷冻干燥器内干燥。干燥后称取适量(精确至0.01g)样品至ASE萃取池,加入50μl(4mg/L)替代物稀释液,放上快速溶剂萃取仪。
1.3 分析条件
1.3.1 ASE萃取条件
载气压力:1.1MPa;加热温度:100℃;萃取池压力:1200psi~2000psi(约合8.3MPa~13.8MPa);预加热平衡:5min;静态萃取时间:5min;溶剂淋洗体积:60%池体积;氮气吹扫时间:60s;静态萃取次数:2次。萃取溶剂根据“2.1萃取条件优化”中的实验选择而定。
1.3.2 氮吹浓缩条件
将氮气开启,使溶剂表面可见气流波动,但要避免形成气涡。氮吹过程中要关注露出的浓缩器管壁,用正己烷多次洗涤管壁,最后浓缩至约1ml。
1.3.3 净化及浓缩条件
将硅酸镁净化小柱固定在固相萃取装置上,用5ml二氯甲烷淋洗净化小柱,加入5mL正己烷,待柱充满后关闭流速控制阀浸润5min,缓慢打开控制阀,继续加入5mL正己烷在填料暴露于空气之前,关闭控制阀,弃去流出液。将浓缩后的提取液转移至小柱中,用0.7mL正己烷洗涤氮吹管,洗液全部转入小柱中,重复三次。缓慢打开控制阀,在填料暴露于空气之前关闭控制阀,加入适量(体积根据“2.2净化条件优化”中的实验选择而定)二氯甲烷-正己烷(2+8)混合溶剂进行洗脱,并收集全部洗脱液。净化后的试液再次按照氮吹浓缩的步骤浓缩至1ml以下,加入适量内标并定容至1.0mL,使内标浓度为200μg/L,混匀后转移至2mL样品瓶中,待测。
1.3.4 气相色谱条件
进样口温度:根据“2.3分析条件优化”中的实验选择而定;不分流;进样量:1.0ul;柱流量:1.0ml/min(恒流);柱温:80℃保持2min,以20℃/min速率升至180℃,保持5min,以10℃/min速率升至290℃,保持10min。
1.3.5 质谱条件
电子轰击源(EI);离子源温度:根据“2.3分析条件优化”中的实验选择而定;离子化能量:70eV;接口温度:280℃;四级杆温度:根据“2.3分析条件优化”中的实验选择而定;溶剂延迟时间:5min;扫描模式:选择离子模式(SIM)模式。
HJ805-2016方法中给出的苯并(a)芘同分异构体定性离子用253、250、251,但实验中发现,253是HP-5MS色谱柱的柱流失离子,不宜使用,故本次实验苯并(a)芘同分异构体定性离子用250、251,其他目标物定性离子与HJ805-2016方法中的一致。
1.4 数据处理
本文实验数据统计、分析利用EXCEL软件和SPSS17.0完成。
2 结果与讨论
2.1 萃取条件优化
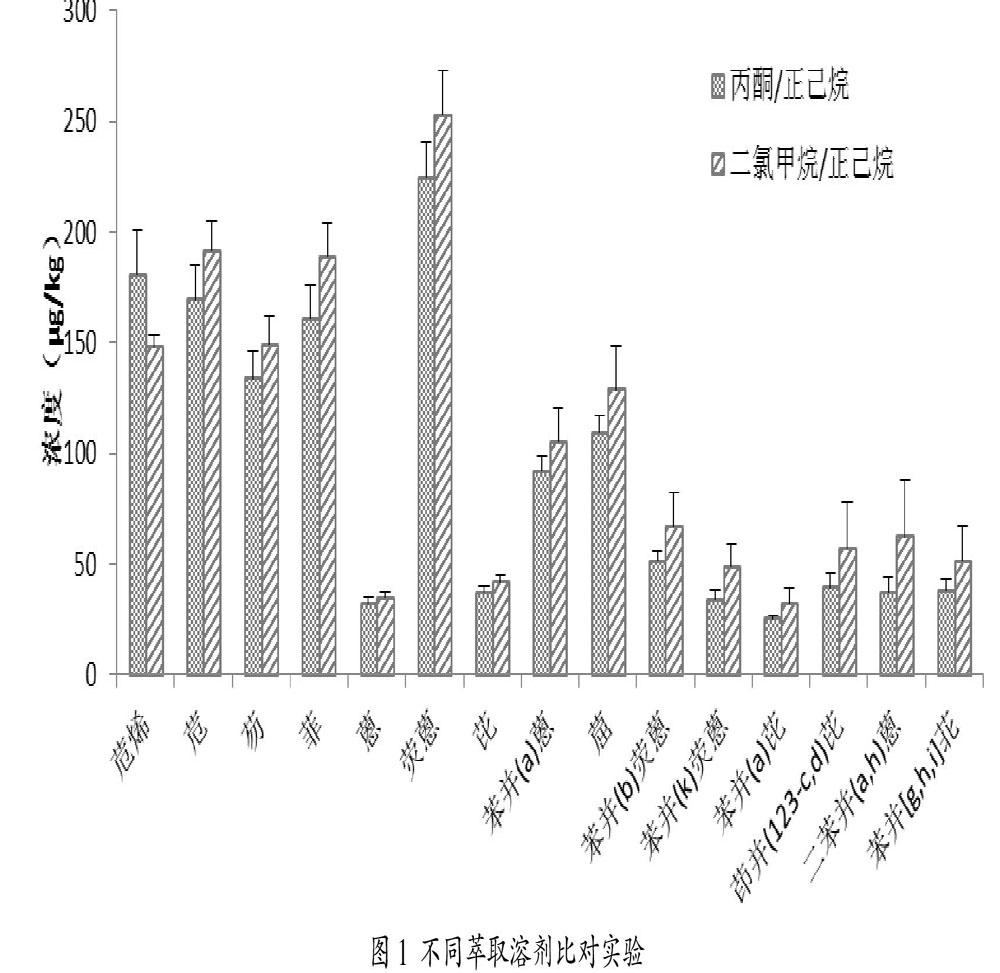
《土壤和沉积物 有机物的提取 加压流体萃取法》(HJ 783-2016)中使用(1+1)丙酮/正己烷为多环芳烃萃取溶剂[10],考虑到丙酮毒性较大,本文尝试用(1+1)二氯甲烷/正己烷为溶剂做比对实验。实验设计如下:对应每种溶剂条件,分别称取3组1.00g土样(依据前期实验结果,该土样多环芳烃含量约为100μg/kg),按照1.3条件进行萃取、测定。以3组数据均值做柱状图,以标准差做误差线,结果如图1所示。
由图1直观看来,对于大部分目标物来说,(1+1)二氯甲烷/正己烷溶剂的萃取效果看上去更优。进一步对所有目标物结果数据依次进行SPSS单因素方差分析,结果显示两种溶剂萃取效果无显著差异。以苯并(a)芘为例,SPSS软件分析结果截图如图2所示,方差齐性检验结果显示显著性大于0.05,认为方差齐性假设成立,进而进行单因素ANOVA方差分析,得出结果显著性p值为0.148,大于0.05表示两组实验结果无显著差异。各目标物不同溶剂萃取结果统计见表1。
上述分析可得出结论:(1+1)丙酮/正己烷混合溶剂与(1+1)二氯甲烷/正己烷混合溶剂在该实验条件下提取效果无显著差异。考虑到丙酮毒性较大,且实验成本更高,本文后续实验选择使用(1+1)二氯甲烷/正己烷混合溶剂作为萃取剂。
2.2 净化条件优化
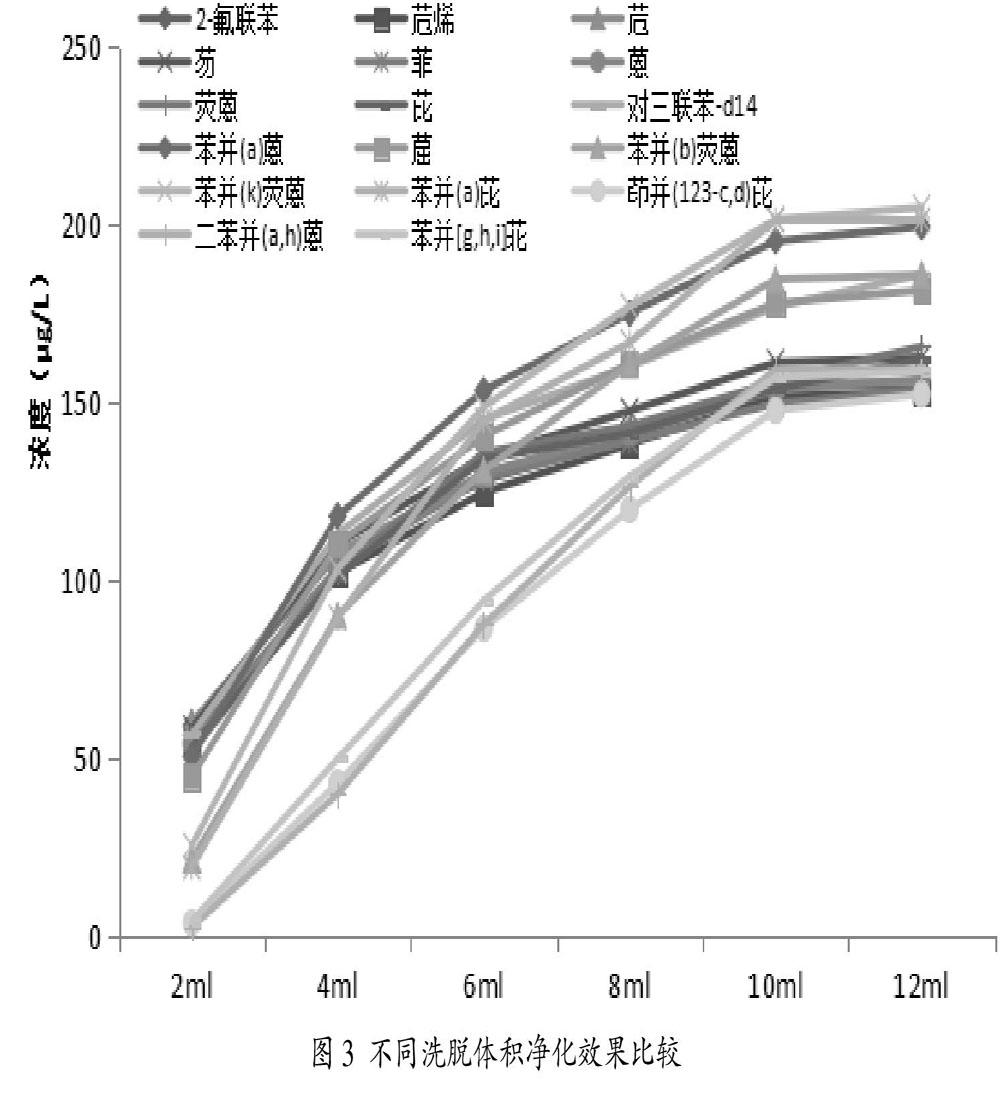
洗脱溶剂和洗脱体积不同,会对净化效果造成影响。本次实验侧重于洗脱剂体积因素影响,实验设计如下:按1.3.3步骤,在6个固定于固相萃取装置上的净化小柱中,分别加入20微升10mg/L的多环芳烃和替代物混标,分别以2ml、4ml、6ml、8ml、10ml、12ml(2+8)二氯甲烷/正己烷溶劑洗脱,收集洗脱液,浓缩,添加内标,转移至进样瓶,上机测定。结果如图3所示。
由图3可知,洗脱溶剂的体积从2ml至10ml梯度递增过程中,对各目标物的洗脱效果不断增强,洗脱体积不宜少于10ml,但10ml和12ml洗脱效果无明显差异。后续实验洗脱条件选择10ml(2+8)二氯甲烷正己烷。
2.3 分析条件优化
2.3.1 质谱扫描模式选择
质谱扫描模式有两种:全扫描SCAN和选择离子SIM模式。其中,HJ805-2016中对应的操作是SCAN全扫描模式,该模式采集的离子多,容易因此造成背景值偏高、干扰大等问题,导致灵敏度无法满足普通土壤样品的微量检测要求。相对而言,SIM模式是对目标物进行特征离子扫描,采集的离子少,具备抗干扰、灵敏度高等优点,在低含量组分定量中有很大优势。但HJ805-2016中对SIM模式没有具体指导。本文采用SIM模式进行测定,对于降低方法检出限,提高检测灵敏度起到了重要作用。
2.3.2 不同温度条件选择
HJ805-2016方法中气相色谱和质谱的温度参考条件为:进样口280℃,四级杆150℃,离子源230℃。本文尝试进行温度条件优化,按1.3.4和1.3.5设定两组温度条件:(1)进样口300℃,四级杆180℃,离子源300℃;(2)进样口280℃,四级杆150℃,离子源230℃。取200μg/L多环芳烃和替代物混标溶液,在上述两种温度条件下分别测定3次,比较响应值,结果如图4所示。
由图4可知,不同温度下,3次进样的重复性均较好。对同一目标物而言,不同温度条件下,响应值的标准偏差接近,但温度较高的条件1响应结果更大,其相对标准偏差更小。所以在该浓度下,认为温度条件1更有利于测定。
2.4 方法性能评价
2.4.1 标准曲线
取8个2ml进样瓶,用微量进样器预先加入适量正己烷溶剂,分别移取适量的多环芳烃标准溶液、替代物标准溶液和内标标准溶液,配制成8个浓度点的校准系列,使得多环芳烃和替代物的质量浓度均分别为5.0μg/L、25.0μg/L、50.0μg/L、100μg/L、200μg/L、500μg/L、1000μg/L,内标质量浓度均为200μg/L。按照前述仪器条件,从低浓度到高浓度依次进样分析。根据HJ805-2016第8.3.1计算平均相对响应因子,同时计算响应因子的相对标准偏差,并绘制校准曲线。结果见表2。
由表2可见,大部分目标物RSD较小,基本在10%以内,二苯并(a,h)蒽RSD相对较大,但仍在HJ805-2016要求的20%范围内。出现此种现象的可能原因有:一是由于仪器型号较老,灵敏度有限造成;二是由于30米柱长仍然不够,对苯并(b)荧蒽/苯并(k)荧蒽以及茚并(123-c,d)芘/二苯并(a,h)蒽的分离能力有限,导致谱图有小部分重叠,定量准确度受到影响。今后可尝试更换其他种类色谱柱或进行仪器改进,进一步提升检测效果。
2.4.2 方法检出限
查阅文献发现,不少同类型研究以仪器检出限代替方法检出限,但是仪器检出限反映的是不同仪器的性能差异,方法检出限才能客观反映样品前处理及测定分析的全过程。本文按照HJ168-2010方法,以石英砂(预先于400℃烘4h)代替土壤样品做7个空白,于ASE萃取前加入的标准物质PAHs稀释液(ρ=100?滋g/L)50?滋L,按照样品分析的全部步骤,重复7次空白加标试验,按MDL=t(n-1,0.99)×SD 计算7份空白加标样品中各个组分浓度的标准偏差(SD)和方法检出限(MDL)。检出限只保留一位有效数字,只进不舍。结果见表3。
表3列举了本实验的方法檢出限和HJ805-2016规定的检出限要求,结果显示,本实验所有目标物检出限均远低于HJ805-2016检出限要求,完全能够满足实际工作需要。
2.4.3 准确度和精密度
精确称量2.00g土样,填充32.0g石英砂,平行制备6份样品。对其中3份样品进行基体加标,加标量为300ng,制备成加标土壤样品。按前述实验方法提取,并上机测定,得到各目标物的相对标准偏差范围2.6%~13.3%,平均加标回收率范围41.7~128%。实验结果满足HJ805-2016中关于“平行样测定结果相对偏差应小于30%,基体加标回收率范围为40%~150%”的质控要求。本实验所有样品都有加替代物,2-氟联苯回收率范围为49.9%~70.8%;对三联苯-d14回收率范围为73.8%~75.8%。各目标物精密度和回收率结果见表4。
3 结论
本研究建立了加压流体萃取-气质联用法用于测定土壤中15种多环芳烃,通过萃取溶剂、净化洗脱、分析温度等条件的对比试验,选择对环境影响相对较小的1:1二氯甲烷/正己烷为ASE萃取溶剂,10ml(2+8)二氯甲烷/正己烷溶剂净化洗脱,分析温度为进样口300℃、四级杆180℃、离子源300℃的优化条件作为方法条件,采用不分流和选择离子模式进行测定。结果表明,此方法在5~1000μg/L范围内,15种多环芳烃相对响应因子的相对标准偏差均小于20%;方法灵敏度高,检出限为0.1~0.4μg/kg,优于现有标准;方法准确度和精密度均满足质控要求,加标回收率和相对标准偏差范围分别为41.7~128%和2.6%~13.3%,符合实际工作中土壤多环芳烃批量痕量检测要求。
参考文献:
[1]王大陆,汪宏杰,王丽娜,等.气相色谱-质谱联用和高效液相色谱法对多环芳烃的分析和对比[J].河南化工,2018,35(5):53-55.
[2]冯小康,朱强.气相色谱-质谱法测定土壤中16种多环芳烃[J].安徽农业科学,2019,47(14):220-223.
[3]李海燕,李楠,于丹丹.微波萃取-气质联用测定土壤中的16种多环芳烃[J].环境监控与预警,2010,2(1):20-23.
[4]张茜,刘潇威,罗铭,等.快速溶剂(ASE)提取、凝胶渗透色谱(GPC)联合固相萃取(SPE)净化,高效液相色谱法测定土壤中的多环芳烃[J].环境化学,2011,30(4):771-777.
[5]中华人民共和国环境保护部.HJ805-2016土壤和沉积物多环芳烃的测定气相色谱质谱法[S].北京:中国环境科学出版社,2016.
[6]中华人民共和国环境保护部.HJ784-2016土壤和沉积物多环芳烃的测定高效液相色谱法[S].北京:中国环境科学出版社,2016.
[7]涂悦鸿,刘细祥.土壤中多环芳烃的预处理及含量分析方法综述[J].广东化工,2010,37(11):258-261.
[8]丁炜炜,杨文清,黄成,等.加压流体萃取-气相色谱质谱法分析水稻土样品中16种多环芳烃的含量[J].广东化工,2018,45(21):101-102.
[9]杨发忠,颜阳,张泽志,等.多环芳烃研究进展[J].云南化工,2005,32(2):44-48.
[10]中华人民共和国环境保护部.HJ783-2016土壤和沉积物有机物的提取加压流体萃取法[S].北京:中国环境科学出版社,2016.
