肺泡蛋白沉积症15例临床分析
2020-01-11莫琼娅潘俊杰董年
莫琼娅 潘俊杰 董年
[摘要] 目的 分析肺泡蛋白沉积症(PAP)的临床表现、辅助检查、诊断方法及治疗效果,以提高临床医生对该疾病的认识。 方法 回顾性分析2005~2019年15例PAP住院患者的临床资料(包括年龄、性别、职业暴露、临床症状及体征、辅助检查、治疗方法等)。 结果 患者临床表现主要为咳嗽、呼吸困难、胸闷,肺功能提示弥散功能下降,影像学检查主要表现为双肺间质性改变,典型特征为“铺路石征”“地图征”改变。确诊主要依靠肺活检或肺泡灌洗液病理明确。9例患者进行全肺灌洗术治疗,1例患者接受雾化吸入治疗。治疗后临床症状均有缓解。 结论 PAP是一种少见的间质性肺病,由于其缺乏特异性的临床表现,在诊断PAP时常常出现误诊漏诊。熟悉掌握PAP的临床表现及影像学特征,可以减少PAP的误诊漏诊,更好地服务临床。
[关键词] 肺泡蛋白沉积症;影像学特征;肺泡灌洗液;全肺灌洗术
[中图分类号] R563.9 [文献标识码] B [文章编号] 1673-9701(2020)31-0040-04
[Abstract] Objective To analyze the clinical manifestations, auxiliary examinations, diagnostic methods and therapeutic efficacies of pulmonary alveolar proteinosis(PAP), so as to improve clinicians' understanding of PAP. Methods The clinical data of 15 cases of PAP inpatients from 2005 to 2019 were analyzed retrospectively, in which the age, gender, occupational exposure,clinical symptoms,physical signs, auxiliary examination,and treatment methods, etc. were included. Results The main clinical manifestations were cough, dyspnea, chest distress, and pulmonary function suggested decreased diffusion function. Imaging examination mainly showed interstitial changes of both lungs, with typical features of "cobble-stone sign" and "map-like" changes. The diagnosis was mainly confirmed by pulmonary biopsy or bronchoalveolar lavage fluid pathology. 9 patients were treated with whole lung lavage, and 1 patient was treated with atomizing inhalation. Clinical symptoms were relieved after treatment. Conclusion PAP is a rare interstitial lung disease. Because of its lack of specific clinical manifestations, it is often misdiagnosed and missed in the diagnosis of PAP. Being familiar with the clinical manifestations and imaging features of PAP can reduce misdiagnosis and missed diagnosis of PAP and better serve the clinic.
[Key words] Pulmonary alveolar proteinosis; Imaging features; Alveolar lavage fluid; Whole lung lavage
肺泡蛋白沉積症(Pulmonary alveolar proteinosis,PAP)是一种以肺泡腔内大量沉积磷脂蛋白样物质为特点的少见肺部弥漫性疾病。每年每百万人口中PAP的发病率和流行率分别为0.36~0.49例和3.7~6.2例[1-2]。PAP患者的临床症状缺乏特异性,但影像学表现尤其是胸部高分辨率CT(High resolution CT,HRCT)有较大的诊断价值[3]。肺活检和纤维支气管镜肺泡灌洗液检查可以明确诊断。PAP患者的临床过程及预后具有个体化差异,部分患者能自行缓解,也有最终发展为呼吸衰竭甚至死亡。
本研究回顾性分析2005~2019年15例PAP患者的临床资料,以期提高对该疾病的认识,为PAP患者的临床管理提供更多信息。
1 资料与方法
1.1一般资料
收集2005~2019年收治的15例PAP住院患者的临床资料。其中男10例,女5例,发病年龄30~68岁,诊断时平均年龄48.4岁。病程5 d~5年,平均病程12.4个月。
1.2方法
回顾性分析15例患者的临床资料,包括年龄、性别、职业暴露史、吸烟史、病程、临床症状、体征、入院时动脉血气分析、血常规、C反应蛋白(C-reactive protein,CRP)、血清乳酸脱氢酶(Lactate dehydrogenase,LDH)、血肿瘤指标、肺功能通气功能及弥散功能检查、胸部CT、治疗方法及治疗效果等,并进行总结与分析。所有患者均通过肺活检和(或)支气管肺泡灌洗液确诊。1例患者经右肺楔形切除术肺活检明确,9例患者经纤维支气管镜下肺泡灌洗液检查明确,2例患者经纤维支气管镜下肺活检明确,3例患者经CT引导下经皮肺穿刺活检明确诊断。
1.3 观察指标
①采集患者空腹外周静脉血5 mL,分离血清,采用Beckman全自动生化分析仪(Au5841)分析生化指标。②采集患者外周静脉血5 mL,使用同舸全自动血细胞分析仪(XE2100)检测血常规白细胞,采用免疫比浊法测定CRP(西门子全自动蛋白分析仪BN Ⅱ System)。③采集患者动脉血3 mL,采用雷度血气分析仪(ABL800)检测血气分析指标,包括动脉血氧分压(PaO2)、肺泡动脉氧分压差[P(A-a)O2)]。④采集患者外周静脉血5 mL,分离血清,置于-20℃冰箱备测,癌胚抗原(Carcinoembryonic antigen,CEA)采用Beckman dxi800酶免疫化学发光法检测,配套试剂盒由Beckman公司提供,神经元特异性烯醇化酶(Neuron specific enolase,NSE)、角质蛋白21-1(Cyfra21-1)采用罗氏E602电化学发光法,采用罗氏配套试剂盒。⑤肺功能:包括第1秒用力呼气容积(Forced expiratory volume for 1 second,FEV1)、用力肺活量(Forced vital capacity,FVC)、第1秒用力呼气容积占用力肺活量百分数(FEV1/FVC%)、肺总量(Total lung capacity,TLC);弥散功能为一氧化碳弥散量占预计值百分比(Carbon monoxide diffusing capacity/predicted value,DLCO/pred%)。⑥胸部CT采用计算机断层扫描系统(SIEMENS SOMATOM Sensation或PHILIPSBRILLIANCE ICT)进行检查。
1.4 统计学方法
采用SPSS 22.0统计学软件进行分析,计量资料以(x±s)表示,组内比较采用配对t检验。P<0.05为差异有统计学意义。
2 结果
2.1 临床特点
7例患者有吸烟史,均为男性。2例患者有粉尘接触史,分别为矿工及木工。最常见的临床症状是进行性加重的呼吸困难11例,咳嗽11例,其次为胸闷7例,发热5例,胸痛2例。2例患者无明显不适症状。体格检查体征缺乏特异性,吸气相爆裂音8例、杵状指6例、发绀3例,4例患者查体无明显异常。
2.2实验室检查
所有患者血常规示白细胞计数均正常。3例患者CRP升高。动脉血气分析结果显示,动脉血氧分压为(66.55±12.58)mmHg,8例患者肺泡动脉氧分压差[P(A-a)O2)]高于40 mmHg,8例患者血清乳酸脱氢酶升高,平均为(288.07±89.50)U/L,9例患者癌胚抗原(CEA)升高,10例患者神经元特异性烯醇化酶(NSE)升高,8例患者角质蛋白21-1(Cyfra21-1)升高。
2.3肺功能检查
所有患者均接受肺功能检查,FEV1/FVC%平均为(86.68±8.96)%,TLC/pred%平均为(69.03±13.69)%,8例患者表现为限制性通气功能障碍,1例患者表现为混合型通气功能障碍,6例患者肺通气功能正常,15例患者均显示DLCO/pred%下降,提示弥散功能下降。
2.4 影像学检查
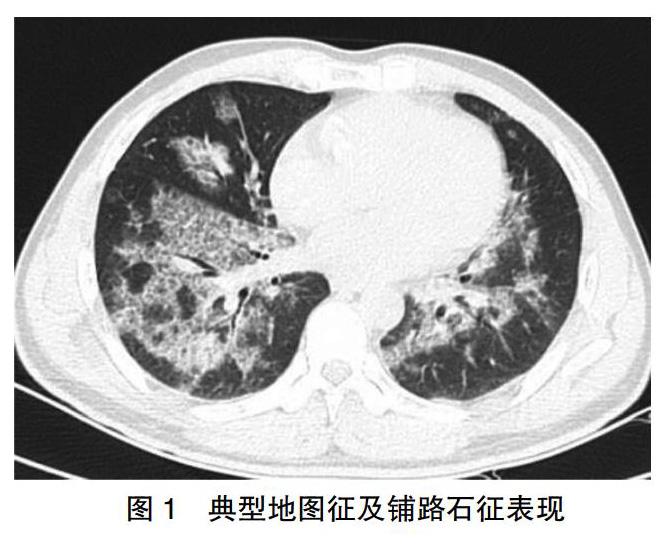
14例患者胸部CT表现为两肺弥漫间质性改变,1例患者表现为单侧右肺病灶。可见典型的“铺路石征”,呈地图状分布。见图1。2例伴有实變,1例伴有曲菌球。
2.5治疗及转归
7例患者接受双侧全肺灌洗术(Whole lung lawage,WLL)治疗,2例患者接受单侧全肺灌洗治疗,1例患者接受粒细胞-巨噬细胞集落刺激因子(Granulocyte macrophage colony stimulating factor,GM-CSF)雾化吸入治疗。
所有接受WLL治疗的患者均有不同程度的临床缓解,或症状好转,或影像学缓解,或肺功能弥散功能改善。7例患者接受WLL治疗前DLCO/pred%为(38.15±10.66)%,治疗后为(47.23±9.59)%,治疗后DLCO/pred%较治疗前升高,差异有统计学意义(t=3.815,P<0.05)。2例患者因缺少治疗后肺功能检查指标,故未纳入。
3 讨论
PAP是一种少见的肺部弥漫性疾病,是由Rosen于1958年首次提出[4],1964年该疾病在我国首次报道,随后逐渐为临床医生所认识。
PAP的病因及发病机制尚未十分明确,普遍认为与肺泡表面活性物质的分泌过多和(或)清除混乱有关。PAP主要有三种类型[5]:①先天性PAP:少见,约占2%,主要发生在婴儿,为常染色体隐形遗传,多与GM-CSF受体链基因突变有关;②自身免疫性PAP(也称为获得性或特发性):最常见,约占90%,认为患者体内存在GM-CSF抗体,导致肺泡巨噬细胞对肺泡表面活性物质清除障碍,大量累积于肺泡腔而致病;③继发性PAP:与基础疾病有关,继发于肺部感染,如结核分枝杆菌、放线菌、肺孢子菌等,或继发于免疫缺陷患者如HIV感染,还可见于血液系统恶性肿瘤淋巴瘤、白血病。另外可见于无机粉尘或化学物质吸入如二氧化硅等。本研究中有2例患者有粉尘吸入病史。
PAP的临床症状缺乏特异性,最常见的症状是进行性加重的劳力性呼吸困难[6]。其次是咳嗽、发热、胸痛及咯血。体格检查阳性体征包括吸气相爆裂音、紫绀,杵状指少见[5]。还可有乏力、食欲减退、体重下降等全身症状。
除非是继发感染,通常实验室检查结果中白细胞计数、C反应蛋白和血沉指标正常。有文献报道,PAP患者LDH[7]、肿瘤指标[8](包括 CEA、cyfra21-1、NSE和表面活性物质相关蛋白 A、B和D[9])可有升高,且其升高程度可能预示着疾病的严重程度[8,10]。本研究15例患者中有多数患者肿瘤指标升高,半数以上患者LDH升高,与相关报道有相似的结果。血气分析可表现为动脉血氧分压下降、低氧血症。本研究中8例患者肺泡-动脉血氧分压差[P(A-a)O2)]>40 mmHg,肺泡-动脉血氧分压差是预测疾病严重程度和功能损伤的较好指标,如肺泡动脉血氧分压差>40 mmHg,则预示患者病情可能会加重进展。
PAP患者最常見的肺功能异常表现为限制性通气功能障碍,一般存在弥散功能下降[2,11]。本研究中所有的病例均存在DLCO/pred%下降,7例患者接受WLL治疗后DLCO/pred%不同程度的升高。WLL后弥散功能得到改善,意味着DLCO/pred%可能可以成为评估治疗效果的指标。
影像学检查在PAP的临床诊断中尤为重要。胸部X线检查常表现为双侧肺门对称性的蝶翼样改变,类似肺水肿但没有心衰的其他表现如心脏增大、胸腔积液等[12]。但胸片表现也可以是不对称、单侧性或局限性。HRCT 可显示更多特征性的征象,典型的表现包括斑片状、磨玻璃样、小叶间隔增厚及小叶内间隔增厚,即所谓的铺路石征[5,12]。尽管铺路石征有时可见于其他间质性肺病[13],其依旧是PAP重要的征象。而且影像学表现与肺功能损害程度有相关性[14]。所有怀疑PAP的患者,除行胸片或普通CT检查外,还应行HRCT检查,以提高诊断的准确性。本研究中有2例患者伴有肺部实变的影像学表现,1例患者为单侧肺病变。
病理诊断是诊断PAP的金标准,光镜下肺泡结构基本正常,肺泡内充满细颗粒状、无结构的过碘酸雪夫(Periodic acid schiff,PAS)染色阳性的蛋白样物质,在终末呼吸性细支气管腔内也可见到[15]。外科开胸肺活检是传统方法,但目前随着纤维支气管镜检查的普及,支气管镜肺泡灌洗及经支气管镜肺活检已成为诊断PAP的主要方法[16]。75%临床疑似病例可以通过乳白色浑浊样肺泡灌洗液中发现大量无定型的PAS阳性的颗粒状物质明确诊断[17]。所以在PAP诊断过程中,关键是能根据临床表现和影像学表现做出判断,做到临床疑诊[5,17],随后进行相关的病理学检查最后明确诊断。在本研究中,所有的病例均是在临床疑诊的基础上最后病理确诊。
目前全肺灌洗仍是主要的治疗方法,主要起缓解作用。一般在全身麻醉下行双腔气管内插管,在支气管镜协助下,一侧肺通气,一侧肺灌洗,反复予37℃生理盐水灌洗,每次灌入1000 mL,回收量丢失不超过150 mL,总灌洗量为10 000~15 000 mL。全肺灌洗的并发症有低氧血症、气胸、继发感染、胸腔积液等。一侧肺灌洗结束后,若无并发症,可于3~7 d后行另一侧肺灌洗。大部分患者经全肺灌洗治疗后症状可得到明显缓解,且能持续较长时间,但少数患者病情仍呈进行性进展,尽管经反复多次全肺灌洗治疗,最终仍死于呼吸衰竭[18-19]。本研究对9例PAP患者进行全肺灌洗术,其中7例患者先后进行双侧全肺灌洗术,2例患者接受单侧全肺灌洗术。在全肺灌洗治疗后患者临床症状或肺弥散功能或CT表现得到不同程度的缓解。根据HRCT随访,有1例患者在全肺灌洗术后3年几乎痊愈,肺部弥漫性病变几乎完全吸收。近年来,GM-CSF的补充治疗逐渐应用于临床。对于部分PAP患者,尤其是GM-CSF缺乏或不足致病的患者,可考虑使用外源性GM-CSF替代治疗。国外研究[20-21]显示GM-CSF吸入治疗原发性PAP有较好的疗效。采用GM-CSF治疗是PAP治疗的新趋势,有待更多的临床研究及临床数据支持。本研究中有1例患者进行GM-CSF雾化吸入治疗,治疗后临床症状好转,但由于缺乏肺功能及胸部CT检查的随访记录,无法从数据上客观评价疗效。其他药物治疗如糖皮质激素治疗效果未得到肯定。
临床上对PAP仍存在误诊漏诊情况,分析原因,由于PAP临床表现缺乏特异性,尤其依靠高分辨胸部CT进行鉴别诊断,医生若对该疾病缺乏认识,对PAP典型的“地图征”“铺路石征”等影像学特征掌握不充足,可能导致误诊漏诊。提高对PAP的诊断意识,熟悉掌握PAP临床诊断的关键线索,将PAP纳入间质性肺疾病或其他表现相似疾病的鉴别诊断中,有助于减少误诊漏诊。
[参考文献]
[1] Inoue Y,Trapnell BC,Tazawar,et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan [J]. American Journal of Respiratory and Critical Care Medicine,2008,177(7):752-762.
[2] Seymour JF,Presneill JJ. Pulmonary alveolar proteinosis:Progress in the first 44 years [J]. American Journal of Respiratory and Critical Care Medicine,2002,166(2):215-235.
[3] IB-D,MJ S. Autoimmune pulmonary alveolar proteinosis:Clinical course and diagnostic criteria[J]. Autoimmunity Reviews,2014,13(4):513-517.
[4] Rosen SH,Castleman B,Liebow AA. Pulmonary alveolar proteinosis[J]. The New England Journal of Medicine,1958, 258(23):1123-1142.
[5] Trapnell BC,Whitsett JA,Nakata K. Pulmonary alveolar proteinosis[J]. The New England Journal of Medicine,2003, 349(26):2527-2539.
[6] Shah PL,Hansell D,Lawson PR,et al. Pulmonary alveolar proteinosis:Clinical aspects and current concepts on pathogenesis[J]. Thorax,2000,55(1):67-77.
[7] Hoffman RM,Rogers RM. Serum and lavage lactate dehydrogenase isoenzymes in pulmonary alveolar proteinosis[J].The American Review of Respiratory Disease,1991,143(1):42-46.
[8] Fang SC,Lu KH,Wang CY,et al. Elevated tumor markers in patients with pulmonary alveolar proteinosis[J]. Clinical Chemistry & Laboratory Medicine Cclm,2013,51(7):1493-1498.
[9] Kuroki Y,Takahashi H,Chiba H,et al. Surfactant proteins A and D: Disease markers[J]. Biochimica et Biophysica Acta,1998,1408(2-3):334-345.
[10] Arai T,Inoue Y,Sugimoto C,et al. CYFRA 21-1 as a disease severity marker for autoimmune pulmonary alveolar proteinosis[J]. Respirology,2014,19(2):246-252.
[11] Goldstein LS,Kavuru MS,Curtis-mccarthy P,et al. Pulmonary alveolar proteinosis:Clinical features and outcomes[J]. Chest,1998,114(5):1357-1362.
[12] Holbert J. CT features of pulmonary alveolar proteinosis [J].American Journal of Roentgenology,2001,176(5):1287-1294.
[13] Johkoh T. Crazy-paving appearance at thin-section CT:Spectrum of diseases and pathologic findings[J]. Radiology,1999,211(1):155-160.
[14] Lee K. Pulmonary alveolar proteinosis:High-resolution CT,chest radiographic,and functional correlations[J]. Chest,1997,111(4):989-995.
[15] Huizar I,Kaburu MS. Alveolar proteinosis syndrome:Pathogenesis,diagnosis,and management[J]. Curr Opin Pulm Med,2009,15(5):491-498.
[16] Suzuki T,Trapnell BC. Pulmonary alveolar proteinosis syndrome[J]. Clinics in Chest Medicine,2016,37(3):431-440.
[17] Wang BM,Stern EJ,Schmidt RA,et al. Diagnosing pulmonary alveolar proteinosis[J]. Chest,1997,111(2):460-466.
[18] Menard KJ. Whole lung lavage in the treatment of pulmonary alveolar proteinosis[J]. Journal of Perianesthesia Nursing,2005,20(2):114-126.
[19] Paschen C,Reiter K,Stanzel F,et al. Therapeutic lung lavages in children and adults[J]. Respiratory Research,2002,6(1):138-148.
[20] Tazawa Ryushi,Trapnell Bruce C,Inoue Yoshikazu,et al. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis[J]. American Journal of Respiratory and Critical Care Medicine,2010, 181(12):1345-1354.
[21] Wylam ME,Ten R,Prakash UBS,et al. Aerosol granulocyte-macrophage colony-stimulating factor for pulmonary alveolar proteinosis[J]. European Respiratory Journal,2006, 27(3):585-593.
(收稿日期:2020-05-14)
