A soluble FcγR homolog inhibits IgM antibody production in ayu spleen cells
2019-10-29KaiChenYuHongShiJiongChenMingYunLi
Kai Chen,Yu-Hong Shi,2,*,Jiong Chen,2,*,Ming-Yun Li
1 Laboratory of Biochemistry and Molecular Biology,School of Marine Sciences,Ningbo University,Ningbo Zhejiang 315832,China
2 State Key Laboratory for Quality and Safety of Agro-products,Ningbo University,Ningbo Zhejiang 315211,China
ABSTRACT Classical Fc receptors (FcRs) mediate the binding to and recognition of the Fc portion of antibodies and play an important role during immune responses in mammals. Although proteins similar to soluble FcRs have been identified in fish, little is known about the role of such proteins in fish immunity. Here, we cloned a cDNA sequence encoding a soluble Fc receptor for an immunoglobulin G (FcγR) homolog from ayu (Plecoglossus altivelis) (PaFcγRl). The predicted protein was composed of two immunoglobulin C2-like domains but lacked a transmembrane segment and a cytoplasmic tail. The PaFcγRl transcripts were distributed at low levels in all tested tissues, but significantly increased after Vibrio anguillarum infection. The PaFcγRl protein was expressed in the head kidney, trunk kidney, and neutrophils. Recombinant PaFcγRl (rPaFcγRl) was secreted when transfected into mammalian cells and the native protein was also detected in serum upon infection. rPaFcγRl was also demonstrated to bind to ayu IgM, as assessed by cell transfection.Suppressive activity of the recombinant mature protein of PaFcγRl (rPaFcγRlm) on in vitro antisheep red blood cell (SRBC) responses was detected by a modified hemolytic plaque forming cell assay. In conclusion, our study revealed that PaFcγRl is closely involved in the negative regulation of IgM production in the ayu spleen.
Keywords: Soluble FcγR homolog; Sequence characterization; IgM-binding protein; Inhibition of IgM production;Plecoglossus altivelis
INTRODUCTION
Antibodies are key components of the immune system, linking both innate and adaptive immunity (Bournazos & Ravetch,2015). The variable Fab region of an antibody mediates specificity and dictates to what antigen and with what affinity the antibody will bind to its target. Antibodies also contain a constant region, termed the Fc domain, which engages diverse cellular receptors, thereby triggering antibodymediated effector functions in innate and adaptive immunity(Dilillo & Ravetch, 2015). As members of the immunoglobulin(Ig) superfamily, Fc receptors (FcRs) are broadly expressed on the surface of various myeloid leukocytes and mediate binding and recognition of the Fc portion of antibodies (Davis,2007). Since their identification three decades ago, our understanding of the biological consequences of FcRs has continued to evolve. FcRs mediate various functions including phagocytosis, antibody-dependent cell-mediated cytotoxicity,lymphocyte proliferation, mast cell degranulation, release/secretion of cytokines and chemokines, antigen presentation,regulation of antibody production, clearance of immune complexes, and Ig transport (Bournazos & Ravetch, 2015;Nayak et al.,2010).
Mammals express six different Ig classes: i.e., IgG, IgM,IgD, IgE, IgA, and IgO. Correspondingly, four major classical FcRs have been identified for IgG (FcγRI, FcγRII, FcγRIII,and FcγRIV), as well as one for IgE (FcεRI), one for both IgM and IgA(FcαμR), one for IgM (FcμR), and one for IgA(FcαRI)in most previously studied placental mammals (Akula et al.,2014). The α chains of these receptors, which are Ig-binding subunits, are all related in structure, thus suggesting an origin from one or a few common early ancestors via successive gene duplications (Davis et al., 2005). In most instances,classical mammalian FcR genes consist of extracellular C2 Ig domains as well as a transmembrane (TM) segment and a cytoplasmic tail (CYT), which may contain signaling motifs(Stafford et al.,2006).
Soluble forms of FcRs (sFcRs) have been identified in the sera and supernatants of activated cells, including T cells, B cells, fibroblasts, macrophages, NK cells, and Langerhans cells (Esposito-Farese et al., 1995). sFcRs are produced either by cleavage at the cell membrane or by secretion of molecules generated by splicing of the TM-encoding exon.Soluble forms exist in the extracellular portion of the receptor or extracellular region linked to the intracytoplasmic portion of the receptor (Rosales & Uribe-Querol, 2013). sFcRs have been described for FcγRI, FcγRII, FcγRIII, FcεRII, and FcαRI(Daëron et al., 1989; Matt et al., 2015; Sarfati et al., 1996; van der Boog et al., 2002). Among these sFcRs, soluble FcγRs(sFcγRs) are relatively well studied and shown to play a regulatory role in immune responses. For example, sFcγRs bind to the Fc domains of IgG to exert immunoregulatory activities in vivo and in vitro, including the inhibition of IgG and IgM antibody production (Fridman et al., 1992) and immune complex precipitation (Gavin et al., 1995). In addition, sFcγRs are also reported to function in the inhibition of C1q binding and complement activation, antibody dependent cellular cytotoxicity, and antigen-antibody uptake (Molenaar et al.,1977).
To date, four classes of Ig have been reported in teleosts,including IgM, IgD, IgZ/IgT, and IgM-IgZ chimera, with IgM reported first (Tian et al., 2009). Of the classical Ig-binding FcR homologs, however, only FcεRIγ has been identified in teleosts, which has limited our understanding not only of the evolutionary history of FcRs but also their functional significance in ectotherms (Akula et al., 2014). Interestingly,FcR homologs without a TM or CYT have been identified in teleosts by genomic/transcriptomic analysis, with the first reported in channel catfish (Ictalurus punctatus) (i.e., IpFcRI)(Stafford et al., 2006). IpFcRI is structurally conserved,containing extracellular domains, as found in classical FcRs,and maintaining three Ig domains and Fc-binding sites for antibody recognition (Stafford et al., 2006). Furthermore,IpFcRI binds to IgM as a soluble protein in serum (Nayak et al., 2010; Stafford et al., 2006). Such proteins are also found in many other bony fish, e.g., zebrafish (Danio rerio), rainbow trout (Oncorhynchus mykiss), tiger puffer (Takifugu rubripes),and spotted green pufferfish (Tetraodon nigroviridis) (Akula et al., 2014; Stafford et al., 2006). To date, however, little is known about the function of these receptors.
Ayu (Plecoglossus altivelis) is an economically important fish in East Asia (Nishimori et al., 2000). Recently, however,the development of ayu aquaculture in China has been severely challenged by Vibrio anguillarum infection, which has resulted in both production and animal welfare problems (Li et al., 2009; Nishimori et al., 2000; Xiong et al., 2018). Given the importance of FcRs in innate immunity, understanding the function and mechanism of action of ayu FcRs would be helpful for disease control and prevention. In this study, a novel sFcγR-like gene (PaFcγRl) was identified from ayu. The mRNA expression profiles of PaFcγRl in healthy and V.anguillarum-infected tissues were determined. The IgMbinding activity of PaFcγRl and its effect on IgM antibody production in spleen cells were also preliminarily characterized.
MATERIALS AND METHODS
Molecular characterization of PaFcγRl
The cDNA sequence of PaFcγRl was obtained from transcriptome data of ayu head kidney-derived monocytes/macrophages (MO/MΦ) using a BLAST search (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The sequence was then used to design primers for specific amplification of the objective gene sequence from cDNA, with the amplicons sequenced on an ABI 3730 automated sequencer (Invitrogen, Shanghai, China)to confirm the correctness of the PaFcγRl sequence. SignalP v4.1 (http://www.cbs.dtu.dk/services/SignalP/) was used to predict the cleavage sites of the signal peptides. The molecular weights (MW) and isoelectric points (pI) were predicted using the Compute pI/Mw tool (http://web.expasy.org/compute_pi/). The N-glycosylation sites were predicted using the NetNGlyc 1.0 server (http://www.cbs.dtu.dk/services/NetNGlyc/). The SMART program (http://smart. emblheidelberg.de/) was used to predict the domain architecture of the putative protein. Multiple sequence alignment was analyzed using ClustalW (http://clustalw.ddbj.nig.ac.jp/), and phylogenetic analyses were conducted using MEGA v7.0(Kumar et al., 2016). Sequences used in this study are listed in Table 1.
Fish and V.anguillarum infection challenge
Healthy fish weighing 50-60 g each were purchased from a commercial farm in Ninghai County, China, and maintained in 100 L tanks at 20-22 °C with regular feeding, as described previously (Chen et al., 2016). The fish were acclimatized to laboratory conditions for two weeks before experiments were conducted. All experiments were performed according to the Experimental Animal Management Law of China and approved by the Animal Ethics Committee of Ningbo University.
The V. anguillarum artificial infection experiment was carried out as reported previously (Chen et al., 2016). Briefly, bacteria were grown in nutrient broth on a rotary shaker at 28 °C andharvested in the logarithmic phase of growth, as monitored by optical density assay. The V. anguillarum cells were washed,resuspended, and diluted to an appropriate concentration in sterile phosphate buffer saline (PBS). Fish were challenged by intraperitoneal injection with 1.2×104colony forming units(CFUs) of live V. anguillarum (in 100 μL PBS) per fish, with PBS alone used as the control. At 4, 8, 12, and 24 h postinfection (hpi), tissue samples were collected and immediately snap-frozen in liquid nitrogen. Blood samples were allowed to clot overnight, after which serum was collected by centrifugation at 13 000 r/min for 15 min at 4 °C. All tissues and sera were preserved at-80°C until subsequent use.
Real-time quantitative polymerase chain reaction (RTqPCR)
Total RNA extraction, first strand cDNA synthesis, and RTqPCR were carried out as reported previously (Ren et al.,2019). The tPaFcγRl(+) and tPaFcγRl(-) primers were used to produce a 227 bp fragment ("t" in the primer name stands for "test") (Table 2). The Pa18S rRNA(+) and Pa18S rRNA(-)primers were used to produce a 116 bp fragment of the housekeeping 18S rRNA gene, which is widely used as an internal control (Table 2). The RT-qPCR protocol was: 94 °C for 5 min,40 cycles of 94°C for 30 s,60°C for 30 s,and 72 °C for 30 s in a RT-CyclerTMreal-time fluorescence quantitative PCR thermocycler (CapitalBio, Beijing, China). The mRNA expression of PaFcγRl was normalized against that of 18S rRNA, and the quantitative differences between expression levels were calculated using the 2-ΔΔCtmethod in the V.anguillarum infection challenge assay (Livak & Schmittgen,2001).All data were analyzed by one-way analysis of variance(ANOVA) with SPSS (v13.0, Chicago, IL, USA). P<0.05 was considered statistically significant.

Table 2 Primers used in the study
Prokaryotic expression and purification of recombinant proteins
In this study, the recombinant mature proteins of PaFcγRl(rPaFcγRlm) and the Fc portion of the ayu IgM (rPaIgM-Fc)were prokaryotically expressed. The primers for amplification of the two genes are listed in Table 2. The prokaryotic expression and purification have been described previously(Zhang et al., 2011). Briefly, amplicons of expected size were digested by EcoR I and Xho I, and subsequently inserted into the pET30a(+) vector. The recombinant plasmids were then transformed into Escherichia coli BL21 (DE3) for overexpression. After overexpression was identified by SDSPAGE analysis, the isopropyl β-D-thiogalactoside (IPTG)-induced cultures were harvested, and the cell pellets were resuspended in 20 mL of sonication buffer. After sonication,the inclusion bodies were recovered and resuspended in 2 mL of buffer A (0.1 mol/L Tris-HCl, 0.5 mol/L NaCl, 10 mmol/L imidazole, and 8 mol/L urea, pH 7.5). The purification of solubilized recombinant proteins was performed on a HisTrapTMFF (GE Healthcare, Shanghai, China) and gradient eluted with buffer B (0.1 mol/L Tris-HCl, 0.5 mol/L NaCl,500 mmol/L Imidazole and 8 mol/L urea, pH 7.5) increasing from 0 to 100%. The peak fractions were pooled and dialyzed with solubilization buffer (50 mmol/L KH2PO4, 1 mmol/L EDTA,and 300 mmol/L KCl, containing 8 mol/L urea and 0.2 mol/L DTT, pH 8.0) and then concentrated using a 10 000 NMWL spin filter (Millipore, Shanghai, China). Refolding of solubilized recombinant proteins by 8 to 2 mol/L urea gradient sizeexclusion chromatography was performed on an XK 16/100 column packed with Superdex 75 gel media (GE Healthcare).The concentrated peak fractions were desalted on a 5 mL Bio-Gel P-6 desalting column (Bio-Rad, Shanghai, China). All procedures were carried out at 4 °C. The purified proteins were lyophilized and stored at-80°C before use.
Antibody preparation and Western blot analysis
Antibody preparation was performed as reported previously(Ren et al., 2019). In brief, purified recombinant proteins,dissolved in PBS and emulsified with Freund's incomplete adjuvant, were used to immunize mice by intraperitoneal injection once every 7 d for a total of four injections. Control mice were injected with incomplete Freund's adjuvant. Whole blood was collected and centrifuged at 13 000 r/min for 15 min at 4 °C to obtain serum. Anti-PaFcγRlm, anti-PaIgM-Fc, and control isotype antisera were purified by protein G chromatography media (Bio-Rad). Antiserum quality was tested by Western blot analysis as described previously (Ren et al., 2019), and visualized using an enhanced chemiluminescence (ECL) kit (Advansta, Menlo Park, USA).The antiserum with 0.2% sodium azide was kept at -20 °C until use. For the determination of PaFcγRl glycosylation,PNGase F (New England Biolabs, Beverly, MA, USA) was used to digest kidney proteins followed by Western blot analysis.
The monocytes/macrophages (MO/MΦ), lymphocytes, and neutrophils were isolated and enriched as described below(Chen et al., 2016, 2018). Ayu head kidney was aseptically extracted and pushed with a glass rod through a 100 μm wire mesh. The resultant single-cell suspension was layered onto Ficoll-Hypaque PREMIUM (1.077 g/mL) (GE Healthcare).After centrifugation at 400 g for 25 min at room temperature,the buffy layer above the Ficoll was collected and washed with RPMI 1640 medium (Invitrogen) supplemented with 2% fetal bovine serum (FBS; Invitrogen), penicillin (100 U/mL),streptomycin (100 U/mL), and heparin (20 U/mL). Cells were cultured for 12 h, with the attached cells identified as enriched MO/MΦ. Blood samples were collected and layered onto Ficoll-Hypaque PREMIUM (1.077 g/mL) at a density gradient,then centrifuged at room temperature for 25 min at 400 g.Neutrophils below the Ficoll (containing erythrocytes) were subjected to hypotonic lysis treatment with ice-cold ACK lysis buffer. The non-adherent lymphocytes were collected from the buffy layer by differential adherence to exclude MO/MΦ.PaFcγRl expression in tissues (liver, spleen, and kidney) and cells (MO/MΦ, lymphocytes, and neutrophils) was determined by Western blot analysis.
In vitro secretion
To test whether the native leader of PaFcγRl could generate a secreted protein, PaFcγRl was amplified using the primers listed in Table 2. The amplified product was cloned into the pcDNA3.1 mammalian expression vector (Invitrogen) and then transiently transfected into HEK293 cells using FuGENE®6 Transfection Reagent (Promega, Madison, USA). The HEK293 cells were grown at 37 °C in a humidified incubator in the presence of 5% CO2. The complete medium for the HEK293 cells was Dulbecco's modified Eagle's medium supplemented with 10% FBS. After 24 h, the supernatants and cell lysates were examined for recombinant PaFcγRl(rPaFcγRl) (containing signal peptide) by Western blotting using the anti-PaFcγRlm antiserum. To further demonstrate the secretion characteristics of PaFcγRl, Western blotting was used to detect the native protein in ayu serum.
Interaction between PaFcγRl and ayu IgM (PaIgM)identified by flow cytometry
The interaction between two recombinant proteins was identified by flow cytometry, as described in Seijsing et al.(2013). Firstly, a recombinant TM protein, with mCherry on its N-terminal and PaIgM heavy chain on its C-terminal (mCherry-TM-PaIgM), was expressed on the HEK293 cell membrane.The TM-PaIgM fragment (111 bp+1 761 bp=1 872 bp) was obtained by overlap extension PCR. The primers for amplifying TM were designed according to the TM domain (aa 53-75) of the ayu C-type lectin receptor gene (Table 2). The control protein (mCherry-TM) was a recombinant TM protein with mCherry on the C-terminal. To construct the recombinant plasmids, amplicons of expected size were digested by BamH I and EcoR I, and subsequently inserted into the pCMV-NmCherry vector (Beyotime Biotechnology, Shanghai, China).The recombinant plasmid was then constructed and transfected into the HEK293 cell line by FuGENE®6 Transfection Reagent. The expression of recombinant mCherry-TM-PaIgM and mCherry-TM were visualized using a Nikon Eclipse Ti-U fluorescence microscope. Their expression levels were also detected using anti-mCherry antibody by Western blot analysis.
The HEK293 cells expressing mCherry-TM-PaIgM or mCherry-TM were incubated with anti-PaIgM-Fc antiserum for 1 h at 37 °C. After washing, the cells were incubated with secondary antibody, FITC-labeled goat anti-mouse IgG(Beyotime Biotechnology), for 1 h at 37 °C. Finally, the cells were washed and analyzed by a Gallios flow cytometer(Beckman Coulter,California,USA).
The HEK293 cells expressing mCherry-TM-PaIgM or mCherry-TM were respectively incubated with rPaFcγRlm for 1 h at 37 °C, and then washed and incubated with anti-PaFcγRlm antiserum for 1 h at 37 °C.After this, the cells were washed and incubated with FITC-labeled goat anti-mouse IgG(Beyotime Biotechnology) for 1 h at 37 °C, then washed and resuspended in PBS supplemented with 1% bovine serum albumin (BSA). Cells were analyzed in a Gallios flow cytometer and 10 000 events per sample were recorded. The data were processed using Kaluza software (Beckman Coulter).
Primary anti-sheep red blood cell (SRBC) responses in vitro
The biological activity of rPaFcγRlm was tested on primary anti-SRBC responses in vitro using a modified hemolytic plaque-forming cell (PFC) assay (Jacobson et al., 2003;Kaattari et al., 1986; Smith, 1998; Varin et al., 1989).Anesthetized fish were sacrificed and placed on ice. The spleens were excised under sterile conditions and immediately placed into 2 mL of cold Hank's balanced salt solution (HBSS) (Sangon Biotech, Shanghai, China) in sterile plastic 2 cm petri dishes. Spleen cells were separated by gentle maceration over a 100 μm nylon screen, transferred to 15 mL conical tubes, and incubated with ACK Lysis Buffer(Beyotime Biotechnology) for 5 min at room temperature. The spleen cells were washed twice with cold HBSS and centrifuged at 2 000 r/min for 10 min at 4 °C. The resulting cellular pellet was resuspended in 2 mL cold complete media(RPMI 1640, 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin). A 50 μL aliquot was removed and combined with 450 μL of 10% trypan blue for lymphocyte differentiation,enumeration, and viability in a hemocytometer at 40×. Each sample was diluted to 8×106cells per mL in complete media,after which 50 μL of 20% SRBCs per mL was added and the sample was cultured for 3 d at 24 °C and 5% CO2. At day 3,purified rPaFcγRlm was added, which was serially diluted(1: 2) (original concentration 5 μg/mL) to a 100 μL final volume, with a sample without rPaFcγRlm used as the control.At day 5, each sample (250 μL) was combined with 0.15 mL of 20% SRBCs and 0.8 mL of 45 °C 0.7% agar solution, then mixed quickly and poured into 5 cm plastic petri dishes and allowed to solidify. Plates were incubated for 2 h with humidity at 24 °C to allow specific lymphocyte recognition of SRBCs and subsequent antibody production and binding. After this,1 mL of 10% complement source serum (diluted by HBSS)was added and the plates were incubated overnight at 24 °C to allow complement-mediated lysis of the SRBCs. Plaques were enumerated and evaluated for approximate size manually via a low-powered Guiguang XTL-400 dissecting microscope with 10× ocular strength. Inhibition >30% was considered significant in view of the variability of this technique.
RESULTS
Cloning and sequence analysis of PaFcγRl
The cDNA sequence of PaFcγRl was 1 113 nucleotides (nts)in length, comprising a 5'-UTR of 79 nts, 3'-UTR of 125 nts,and open reading frame of 909 nts, and encoding a polypeptide of 302 amino acids (aa) with a predicted molecular weight (MW) of 34.1 kDa. BLAST searching revealed that this sequence was homologous to mammalian FcγR and was thus tentatively named PaFcγRl. It was predicted to comprise of a signal peptide (aa 1-33) and two Ig domains: Ig domain 1 (D1) consisting of 80 aa (aa 43-122)and Ig domain 2 (D2) consisting of 85 aa (aa 130-214). Both Ig domains contained two conserved cysteine residues (Cys),essential for the formation of disulfide bonds (predicted as Cys58-Cys98and Cys145-Cys191) (Figure 1). The two disulfide bonds were conserved in all selected mammalian and fish FcRs (Figure 1B). According to the features, such as the spacing of cysteine bridges and number of beta sheets, D1 and D2 were classified as C2 domains and shared homology with low-affinity receptor domains. Furthermore, PaFcγRl had three potential N-glycosylation sites: i.e., N112NS, N177LT, and N180LT.
Sequence comparison analysis showed that PaFcγRl was tightly grouped with previously reported Nile tilapia(Oreochromis niloticus) (identity 35.4%) and Japanese ricefish(Oryzias latipes) (identity 34.4%), suggesting an evolutionary relationship. Phylogenetic tree analysis revealed that PaFcγRl fell into a larger cluster that included fish FcRs, and also grouped with the mammalian FcγR subset (Figure 2A). In addition, each PaFcγRl domain had a related counterpart that variously occurred throughout the FcγRs(Figure 2B).
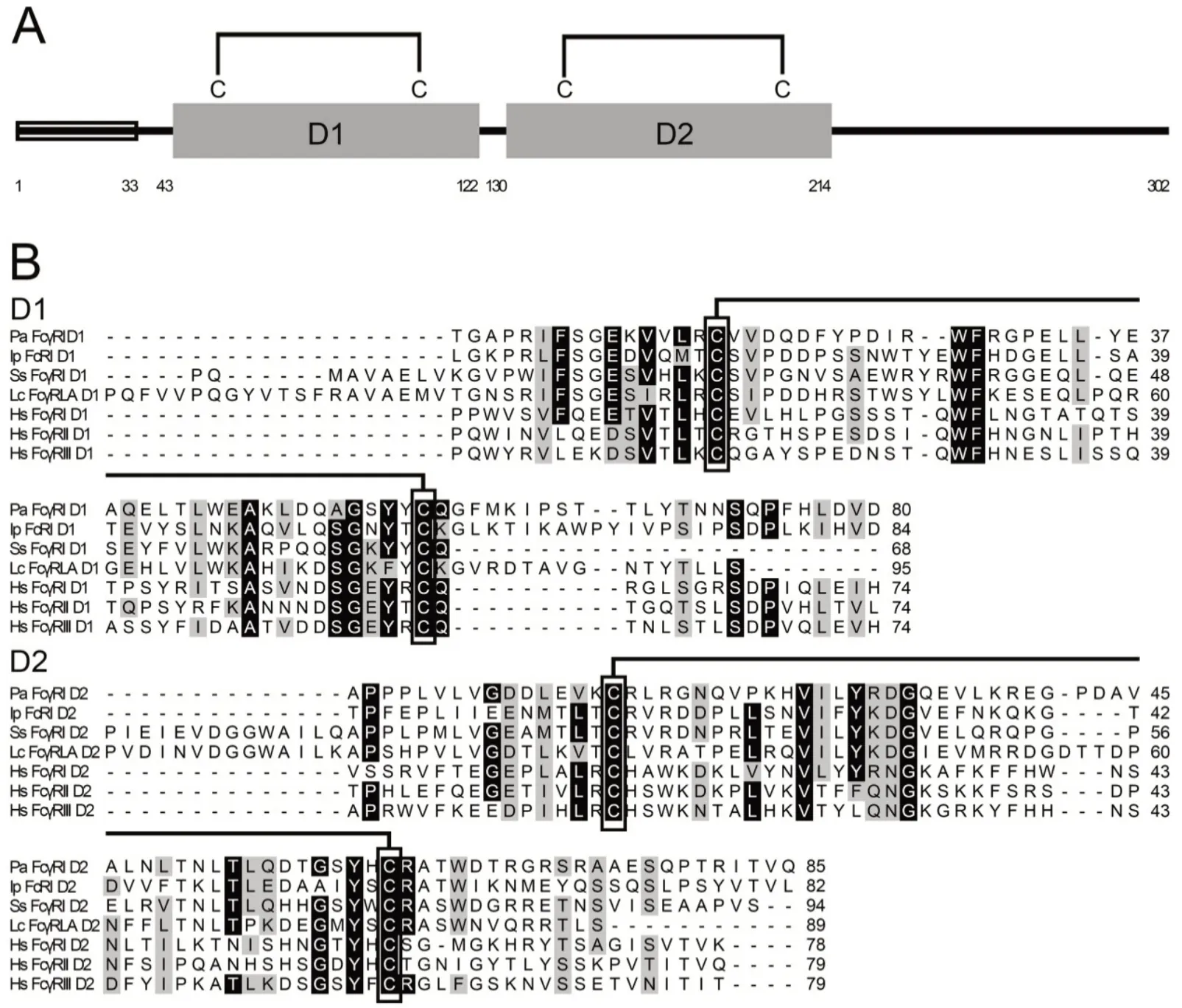
Figure 1 Schematic representation and multiple alignments of amino acid sequences

Figure 2 Phylogenetic analysis of full-length (A) and two Ig domain (B) amino acid sequences of PaFcγRl and related FcRs using neighbor-joining method
Tissue expression profiles of PaFcγRl transcripts
PaFcγRl mRNA was expressed in the liver, spleen, kidney,brain, intestines, muscle, and gill at low levels, with the highest expression detected in the gill of healthy ayu (Figure 3A). Following V. anguillarum infection, PaFcγRl mRNA expression was up-regulated in all tested tissues (Figure 3BE). After stimulation with viable V. anguillarum, the upregulation of PaFcγRl expression in the liver and spleen demonstrated an inverted U-shaped tendency. The highest expression levels of PaFcγRl in the liver, spleen, and kidney were detected at 8 hpi and in the gill were detected at 4 hpi.Furthermore, PaFcγRl mRNA expression levels in the liver,spleen,kidney,and gill were up-regulated by 19.30-fold,16.05-fold, 19.01-fold, and 4.78-fold, respectively, after stimulation(Figure 3B-E).
Prokaryotic expression, purification, and antibody preparation of rPaFcγRlm and rPaIgM-Fc
After induction by IPTG, the recombinant proteins were all over-expressed in E. coli BL21 (DE3). The size of rPaFcγRlm on the 12% SDS-PAGE gel was ~40 kDa, similar to the calculated MW of 39.8kDa (34.1 kDa PaFcγRlm plus 5.7 kDa His-tag) (Figure 4A). The size of rPaIgM-Fc on the 12% SDSPAGE gel was 44 kDa, similar to the calculated MW of 43.2 kDa (37.5 kDa rPaIgM-Fc plus 5.7 kDa His-tag) (Figure 4C). The purifications of the two recombinant proteins were all greater than 95%.
Western blot analysis revealed that the MW of native PaFcγRl in the ayu kidney was ~47 kDa and after deglycosylation was ~34 kDa, similar to sequence calculations(34.1 kDa) (Figure 4B). Western blot analysis also revealed that the MW of the native PaIgM heavy chains in ayu was ~70-80 kDa, similar to the MW of IgM heavy chains reported previously in fish(Wilson&Warr,1992)(Figure 4D).
Furthermore, Western blot analysis revealed native protein expression in the healthy head kidney and trunk kidney, but not in the liver or spleen (Figure 4E). The detected cell types that expressed PaFcγRl were neutrophils, but not MO/MΦ or lymphocytes(Figure 4E).
In vitro secretion of rPaFcγRl
Results showed rPaFcγRl with the native leader was expressed in the HEK293 cells. As shown in Figure 5A,rPaFcγRl was ~49 kDa (calculated MW 34.1 kDa), which might be caused by glycosylation, as expressed in HEK293 cells. rPaFcγRl was detected in both the supernatants and cell lysates of pcDNA3.1-PaFcγRl-transfected cells but not in the supernatants and cell lysates of the control cells transfected with pcDNA3.1, indicating that the native leader can indeed generate a secretory PaFcγRl. Native PaFcγRl in serum was also detected by Western blotting. In the sera of the healthy control and infected ayu, no obvious bands were observed at 4, 8, or 12 hpi, whereas an obvious band was observed in the infected ayu at 24 hpi(Figure 5B).
rPaFcγRlm binding to PaIgM
PaIgM was expressed as a fusion protein, which included PaIgM+TM+mCherry. Its expression was first detected in HEK293 cells by fluorescence microscopy. Results showed that mCherry was expressed in the cytoplasm, and the two recombinant proteins (mCherry-TM and mCherry-TM-PaIgM)were mainly localized on the plasma membrane (Figure 6A).Western blot analysis revealed that the MW of mCherry-TM was ~31 kDa, similar to the calculated MW of 30.5 kDa(3.8 kDa TM plus 26.7 kDa mCherry),and the MW of mCherry-TM-PaIgM was ~95 kDa, similar to the calculated MW of 95 kDa (68.3 kDa TM-PaIgM plus 26.7 kDa mCherry) (Figure 6B). Flow cytometry revealed that PaIgM was identified with anti-PaIgM-Fc antiserum, suggesting that the PaIgM part in mCherry-TM-PaIgM was mainly localized outside the HEK293 plasma membrane (Figure 6C). Finally, the interaction between rPaFcγRlm and the PaIgM Fc portion was detected by flow cytometry. Results showed that cells probed with FITC displayed a linear relationship between mCherry and FITC fluorescence, indicating a linear correlation between the amounts of mCherry-TM-PaIgM and binding of rPaFcγRlm(Figure 6D). The control (mCherry-TM and rPaFcγRlm) did not show such a correlation(Figure 6D).
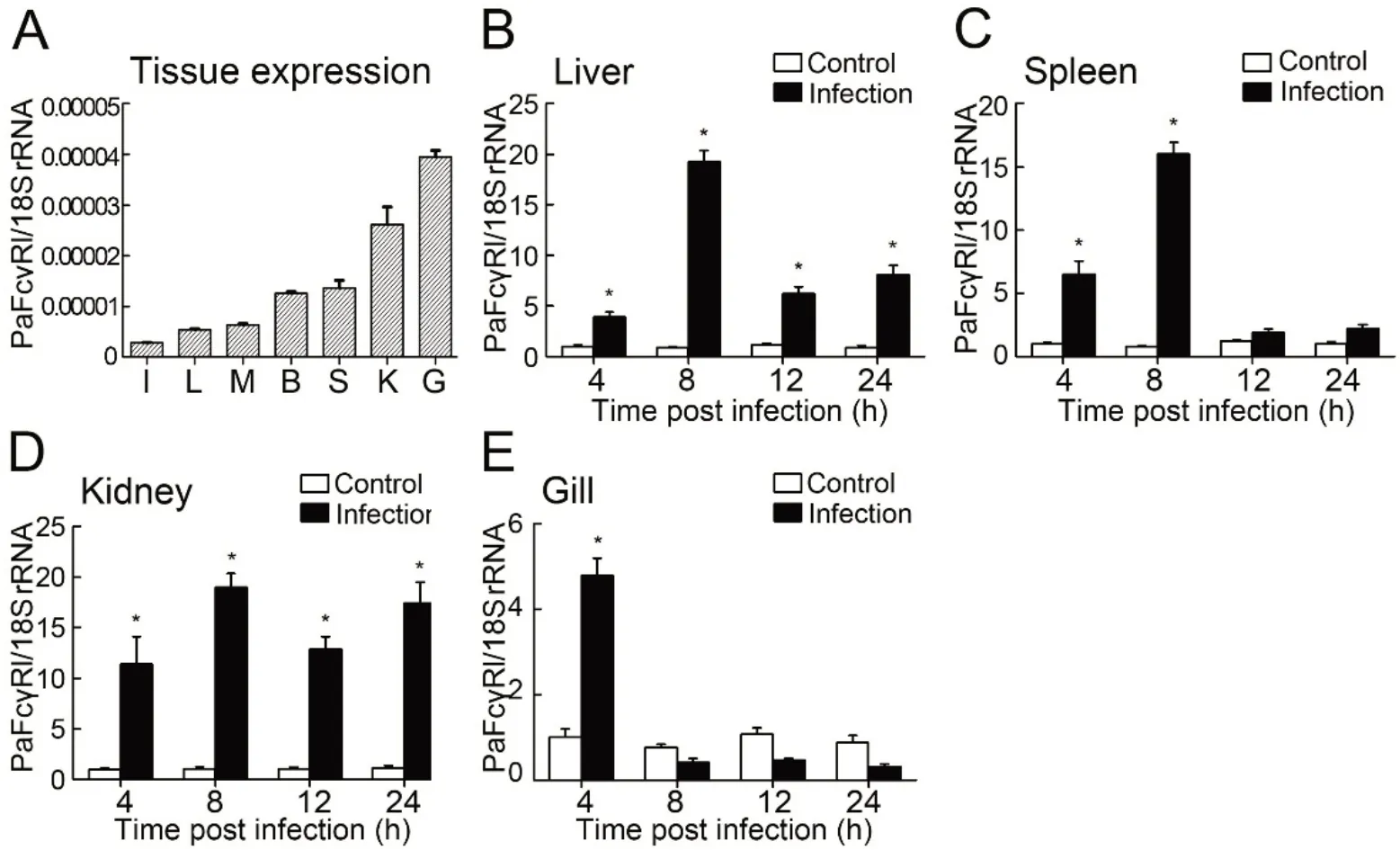
Figure 3 RT-qPCR detection of PaFcγRl expression in ayu tissues
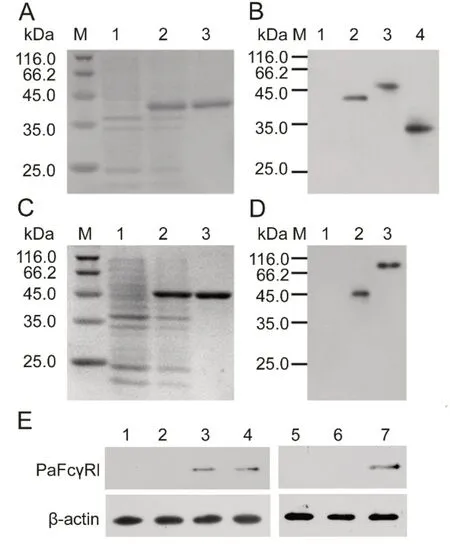
Figure 4 Prokaryotic expression and Western blot analysis
Suppressive activity of rPaFcγRlm on in vitro IgM anti-SRBC antibody responses
The biological activity of rPaFcγRlm on primary anti-SRBC antibody responses in vitro was tested. As shown in Figure 7,rPaFcγRlm suppressed anti-SRBC antibody production in a dose-dependent fashion. About 1.70 μg/mL of rPaFcγRlm was necessary to inhibit 50% of antibody production against SRBCs. This effect was not due to toxicity, as the addition of rPaFcγRlm did not decrease viability of the spleen cells.

Figure 5 Secretion of rPaFcγRl
DISCUSSION
Myeloid leukocytes in humans and mice are equipped with a variety of receptors that enable their interaction with monomeric or aggregated immunoglobulins, antigen-antibody immune complexes, and opsonized antibody-coated particles or cells (Pierre & Friederike, 2015). FcγRs are receptors that induce diverse biological functions after binding to the Fc portion of antibodies mediated by extracellular domains. The extracellular domains of mammalian FcγRI contain three Ig domains (i.e., D1, D2, and D3), whereas FcγRII, FcγRIII, and FcγRIV only contain the first two (i.e., D1 and D2). D1 and D2 are highly homologous among species, whereas D3 shows a lower level of homology, although it is still clearly related to the Ig superfamily (Sears et al., 1990). Interestingly, there is a group of teleost proteins closely homologous to the extracellular domains of mammalian FcγRs (Stafford et al.,2006).Among such homologous proteins, most contain two Ig domains (D1, D2), although a few contain three or four. For example, IpFcRI contains three Ig domains (D1, D2, and D3),with D1 and D2 found clustered with high bootstrap values with mammalian FcR Ig-domain counterparts, but D3 not phylogenetically related to any mammalian FcR Ig domain(Stafford et al., 2006). Likewise, there are a different number of Ig domains in the FcR sequences of zebrafish, rainbow trout, tiger puffer, and spotted green pufferfish (Akula et al.,2014; Stafford et al., 2006). Here, we studied an FcγR-like gene, PaFcγRl, from ayu. Phylogenetic tree analysis showed that PaFcγRl and other fish FcRs were grouped together and were also clustered with the mammalian FcγR subset. In addition, PaFcγRl possessed only D1 and D2, which were classified into C2 sets and phylogenetically related to the corresponding domain of mammalian and fish FcγRs,respectively.
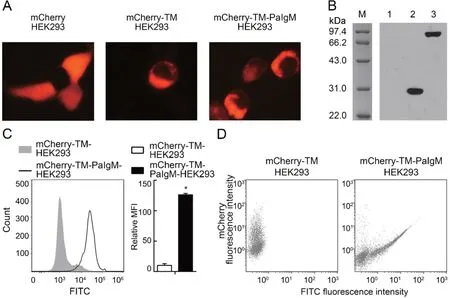
Figure 6 Interaction between rPaFcγRlm and PaIgM

Figure 7 rPaFcγRlm inhibits anti-SRBC antibody production
Tissue and cellular mRNA expression of the FcγR homolog has been studied in channel catfish. Stafford et al. (2006)readily detected IpFcRI transcripts by Northern blot analysis in the spleen and hemopoietic kidney tissues of channel catfish,as well as in their peripheral blood leukocytes (PBLs)(predominantly in granulocytes). To date, however, no information regarding mRNA expression of FcγR homologs related to infection has been reported. Mammalian sFcγRs have been detected in biological fluids of mice and humans,with production found to be highly dependent on environmental factors, including pathogens. For example,mouse sFcγRII (Fcγ2b/γ1R) was reported to increase in culture medium after stimulation of spleen cells with lipopolysaccharide (LPS), which was attributed to B lymphocytes (Pure et al., 1984). Furthermore, a dramatic increase in circulating sFcγRII is also found in Schistosoma mansoni-infected mice (Khayat et al., 1986). In the current study, PaFcγRl mRNA was expressed at low levels in all tested tissues, with the highest expression found in the gill.Furthermore, expression in the liver, spleen, and kidney showed sensitive responses to V. anguillarum infection. With antiserum, the PaFcγRl protein was also detected in the head kidney,trunk kidney,and neutrophils.
The teleost proteins mentioned in this study are soluble,thus differing from the mammalian membrane-bound FcγRs.This suggests that this group of proteins may be secreted and/or intracellularly expressed, thus hinting at their function. In channel catfish, IpFcRI was identified as a secretory protein in the serum akin to sFcR found in mammals (Stafford et al.,2006). In this study, rPaFcγRl was also found to be secreted in the supernatants of HEK293 cells. Native PaFcγRl was also detectable in serum challenged with V.anguillarum.
The binding of the Fc region of IgG to FcγR is a critical step for the initiation and control of effector immune functions(Sibéril et al., 2007). For the IgG ligands, FcγRI and FcγRIV are high-affinity receptors, whereas FcγRII and FcγRIII are low-affinity receptors (Qiu et al., 1990). Mouse and human sFcγRs bind to IgG subclasses with a binding profile identical to the corresponding membrane-associated receptors and exhibit immunomodulatory properties (Lynch et al., 1992). In fish, only IpFcRI has been identified to bind with IgM, as assessed by co-immunoprecipitation and cell transfection studies (Stafford et al., 2006). In our research, rPaFcγRlm was found to bind with IgM, which might provide a hint to its function in immunity.
As reported earlier, mammalian sFcγRs inhibit in vitro and in vivo immune responses. For instance, purified mouse sFcγRIIB1 exerts dose-dependent suppressive activity on primary and secondary antibody responses when added to cultures of spleen cells stimulated with SRBCs, with the effect even more pronounced on IgG than on IgM responses (Varin et al., 1989). Sautès et al. (1992) also found that sFcγRIIB1 inhibits antibody in vitro responses to SRBCs in small B cell cultures stimulated by anti-IgM antibodies in the presence of IL-4 and IL-5. Moreover, intraperitoneal injection of this material into adult mice immunized with SRBCs decreases IgG antibody production in spleen cells, as measured by PFC assay, and in serum, as measured by antigen-specific ELISA(Sautès et al., 1992). Furthermore, purified human sFcγRIIIB(sCD16) inhibits IgM and IgG production of human peripheral blood mononuclear cells stimulated by pokeweed mitogen in vitro in a time and dose-dependent manner (Teillaud et al.,1993). sFcγRs are believed to function by competing with their membrane-bound counterparts for Ig (or immune complex) binding, which, in turn, down-regulates B cell proliferation and antibody production (Fridman et al., 1993). In our study, the PaFcγRl protein was not detectable in the serum of healthy ayu but was up-regulated upon infection. We identified that rPaFcγRlm exerts suppressive activity on primary antibody responses in vitro at a relatively high concentration. Thus, we speculated that PaFcγRl might be involved in the down-regulation of antibody levels at the late stage of infection.
In summary, we showed that PaFcγRl exhibited sequence characteristics similar to classical FcRs with extracellular domains. Furthermore, we revealed that PaFcγRl was a secretory protein bound to PaIgM. In vitro, PaFcγRl likely plays a role in the regulation of IgM production.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS'CONTRIBUTIONS
Y.H.S. and J.C. drafted the experiments; K.C. and Y.H.S. performed the experiments; K.C.,Y.H.S., J.C., and M.Y.L. analyzed the data and wrote the paper.All authors read and approved the final version of the manuscript.
杂志排行

Zoological Research的其它文章
- Progress in in vitro culture and gene editing of porcine spermatogonial stem cells
- Counting stripes:revision of the Lipinia vittigera complex(Reptilia,Squamata,Scincidae)with description of two new species from Indochina
- Synchronization between frontal eye field and area V4 during free-gaze visual search
- Immunodetection of ephrin receptors in the regenerating tail of the lizard Podarcis muralis suggests stimulation of differentiation and muscle segmentation
- Genetic diversity and temporal changes of an endemic cyprinid fish species,Ancherythroculter nigrocauda,from the upper reaches of Yangtze River
- Non-invasive genetic analysis indicates low population connectivity in vulnerable Chinese gorals:concerns for segregated population management