钨中不同构型的双自间隙原子扩散行为研究*
2019-07-11冉琴王欢钟睿伍建春邹宇汪俊
冉琴 王欢 钟睿 伍建春 邹宇 汪俊
(四川大学原子核科学技术研究所,辐射物理及技术教育部重点实验室,成都 610064)
1 引 言
钨因其具有高熔点、低溅射产额、高热导率等特性而被视为聚变堆中非常具有应用前景的面向等离子体候选材料[1].这些材料处于严苛的聚变环境,将受到中子和氦氢同位素的轰击,产生空位、自间隙原子 (self-interstitial atoms,SIAs)以及位错等缺陷.这些缺陷的扩散、聚集及演化将导致材料宏观性能的改变(如: 肿胀和变形),最终会影响到装置的安全性问题[2−6].不同于空位,SIAs 及其团簇具有快速的移动性,其扩散行为被认为是决定材料微观性能的关键因素[7−10].其扩散系数及扩散机制等方面的精确知识是我们能正确理解和预测材料微观演化行为的必要条件.因此,钨中SIAs的动力学行为研究至关重要.
目前,钨中SIAs及其团簇的相关特性已经得到了广泛研究[11−17].分子动力学模拟和第一性原理计算表明钨中单自间隙原子(single selfinterstitial atom,1-SIA)最稳定的构型是结构[11,12,18].Tsong和Casanova[13]通过场粒子显微镜观察到钨中1-SIA与双自间隙原子(di-interstitial atoms,2-SIAs)团簇沿着方向的扩散.Amino等[14]利用透射电子显微镜观察了纳米级SIAs团簇的形成过程,并用动力学蒙特卡罗研究了团簇的扩散行为.他们得出低温下钨中SIAs扩散是一维运动,这些实验结果都与第一性原理[1]和分子动力学[4,15]研究结果一致.值得一提的是,同为体心立方 (body center cubic,BCC)晶体结构的Fe中SIAs的扩散却是三维运动,这表明在BCC金属中存在不同的SIAs扩散机制.Zhou等[5,15]利用分子动力学方法研究了钨中SIAs团簇的形成和运动特性,结果表明2-SIAs形成的团簇在较低温度下非常稳定,即使在900 K也未发生转向,保持一维运动,SIAs团簇的迁移能与团簇尺寸大小无关.最近,Swinburne 等[19]用密度泛函理论结合量子分子动力学方法研究了金属钨中SIAs在低温下的扩散行为,研究表明SIAs在10 K左右的温度下也具有快速的迁移行为.尽管有大量文献研究金属钨中SIAs,但对钨中具有不同构型SIAs团簇的扩散行为鲜见报道,对其扩散机理仍有待研究.分子动力学不仅能在原子尺度模拟缺陷的微观演化行为,揭示扩散机理,而且在多尺度模拟中起着重要的作用.它能为动力学蒙特卡罗模拟提供输入参数(如: 扩散系数和扩散机制),从而进行长时间、大空间尺度的模拟以获得实验上可观察的结果,进而解释和预测实验.2-SIAs作为一种重要的间隙团簇生长核,是大尺寸SIAs团簇的生长基础,研究其不同构型无论是对理论模拟还是实验研究都有一定的指导意义.
本文基于新发展的钨原子间相互作用势,采用分子动力学方法研究了钨中SIAs的扩散行为.主要考察了具有不同构型的2-SIAs随温度变化的扩散行为.通过将微动弹性带 (nudged elastic band,NEB)算法获得的SIAs迁移能与阿伦尼乌斯(Arrhenius)拟合的结果进行对比,阐明了钨中1-SIA与2-SIAs的扩散系数随温度的变化规律.
2 模型与方法
分子动力学模拟的核心问题是原子间作用势的选取,它直接关系到能否准确描述原子间的相互作用以及真实可靠地反映所模拟的物理过程.Bonny等[20]根据实验和密度泛函理论计算结果,详细评估了19种钨势的基本物理属性,特别是点缺陷和位错属性.结果表明,Marinica 等[21]发展的EAM2势总体性能最佳.因此,本文也采用EAM2势,它是根据钨晶体的晶格常数、弹性常数、内聚能、点缺陷形成能等物理参数和来自于第一性原理的原子间作用力数据拟合而成,形式如下:
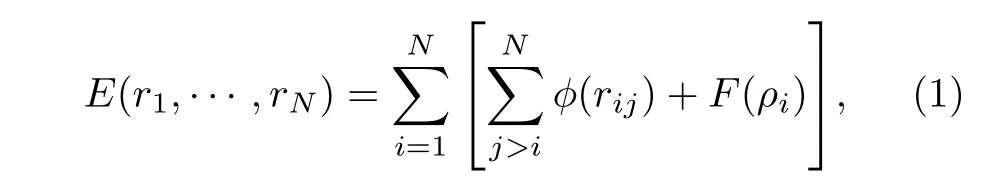
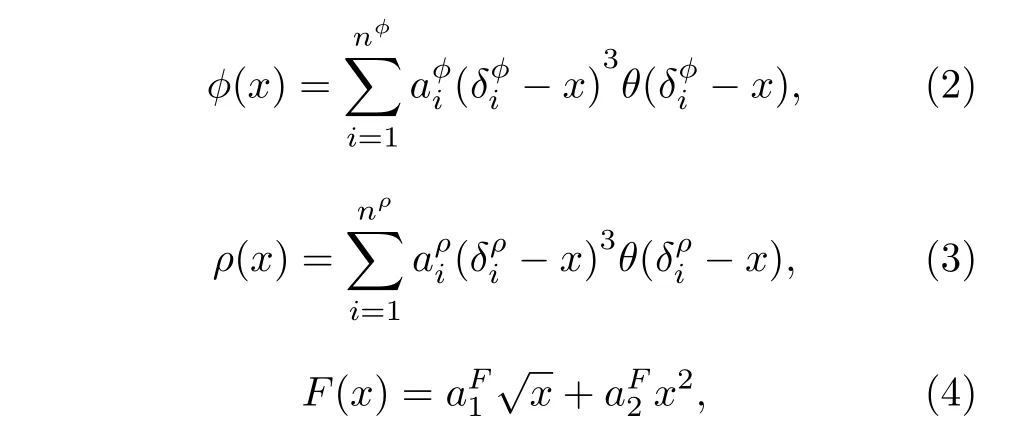
式中 ϕ 为两体作用函数; ρ 为有效电子密度;F 为嵌入函数; θ(x) 是 Heaviside 阶跃函数,当时,其他情况下上述参数的具体取值见文献[21].
在进行分子动力学模拟之前,首先构建一个BCC 结构的钨晶体,基底共 20 层,每层 10 × 10个原子排列在(001)面.根据研究的具体需要,引入SIAs.本文所涉及的SIAs结构包括1-SIA (图1)和多种构型的 2-SIAs (图 2),2-SIAs结构包括最近邻(2-SIAs-1st)、次近邻(2-SIAs-2nd)、三近邻结构(2-SIAs-3rd)以及非平行结构.为了将体系扩展到无限大情形,分别沿着 [100],[010],[001]方向加上周期性边界条件.值得一提的是,当SIAs运动出基底盒子时,由于周期性边界条件的作用,SIAs将会从另外一面进入基底,这将会丢失掉真实的扩散信息.为了获得SIAs在无限大体系中的运动轨迹,在SIAs扩散出基底时修正了其位置信息,具体的修正方式见文献 [22,23].本文所有的模拟都在NVT (固定粒子数、体积、温度)系综下进行,为了将系统升至所需温度,将系统中的每个原子按Maxwell随机分配速度,然后弛豫.在演化过程中利用 Wigner-Seitz cell方法判断 SIAs的位置,并按一定的时间间隔保存位置信息,从而得到SIAs 运动的轨迹.演化的时间步长设为 10–15s (1 fs),每一种结构配置演化时间为10 ns.
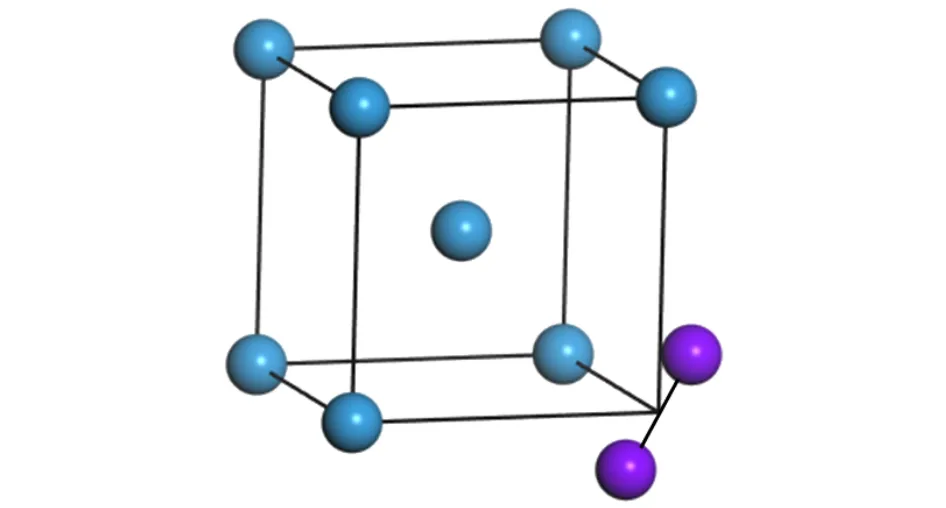
图1 钨中 1-SIA 的结构图 (紫色球为 SIAs结构,蓝色球为格点原子)Fig.1.1-SIA configuration in W.The purple sphere represents the SIA;the blue one represents the lattice atom.
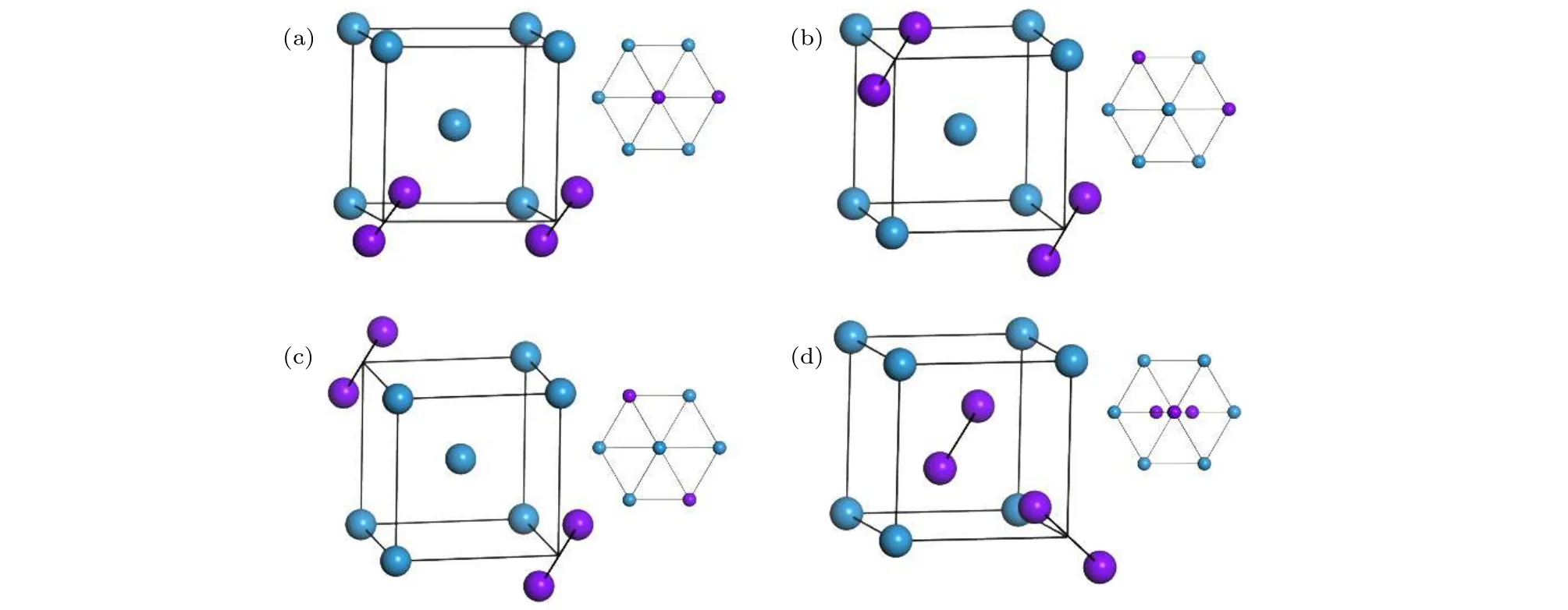
图2 2-SIAs的不同构型图 (a),(b),(c),(d)分别代表最近邻、次近邻、三近邻以及非平行结构的结构示意图;右上方的插图分别代表这几种结构方向的视图;紫色球为SIAs,蓝色球为格点原子Fig.2.Schematic illustrations of the 2-SIAs with different configurations: (a),(b),(c),(d) Represent the configuration of the 2-SIAs-1st,2-SIAs-2nd,2-SIAs-3rd and the non-parallel SIAs,respectively.Insets represent the views corresponding to their orientations;the purple sphere stands for the SIA and the blue one stands for the lattice atom.
3 结果与讨论
3.1 形成能与束缚能
为了考察各种SIAs结构的相对稳定性,首先从能量角度考察了其形成能和束缚能.重点研究了2-SIAs的平行结构和非平行结构,平行结构包括图2(a)—(c)所示的最近邻(2-SIAs-1st)、次近邻 (2-SIAs-2nd)、三近邻结构 (2-SIAs-3rd);非平行结构如图2(d)所示.SIAs的形成能为
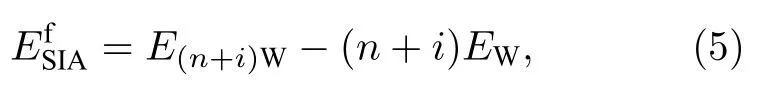
2-SIAs的束缚能公式:
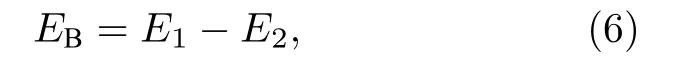
其中 EB为束缚能,E1为 SIAs 之间无相互作用的系统能量,E2代表两者结合形成稳定结构时系统的最低能量.形成能和束缚能的计算结果,分别见表1和表2.通过计算得到单个SIAs的形成能为 10.406 eV,这与文献 [20]结果吻合.可以看出,对于2-SIAs,最近邻结构具有最低的形成能(18.574 eV),同时具有最高的束缚能 (2.245 eV),是最稳定的结构.有趣的是,非平行结构的稳定性仅次于最近邻结构,而高于次近邻和三近邻结构,最不稳定的结构是三近邻结构.这些结论与本文随后的动力学模拟结果一致.

表1 不同缺陷结构的形成能Table 1.Formation energies of self-interstitials with different configurations.

表2 不同缺陷结构的束缚能Table 2.Binding energies of self-interstitials with different configurations.
3.2 单个自间隙原子的扩散
在BCC金属中,基于不同的材料有两种最稳定的自间隙结构,一种是结构,另一种是结构.金属铁中的最稳定结构是前一种[5],而钨中是后一种[11,12].两种不同的稳定结构导致它们呈现出不同的扩散行为.为了了解钨中1-SIAs的扩散行为,考察了钨中结构的1-SIA在100—1500 K温度范围内的扩散,并采用Wigner-Seitz cell方法得到其扩散径迹(图3).结果表明:1-SIA 在 600 K 以下,沿着方向表现为快速的一维运动.这与分子动力学[4,5]和第一性原理计算[1]低温下的扩散结果一致.当温度达到700 K时,SIA的运动方向会随机转向其他三个等价的方向,从而呈现三维运动形式.如图3所示,随温度升高,SIA 转向越频繁.需要注意的是,对于不同的势函数,钨中SIAs的旋转温度不同[22].

图3 1-SIAs在不同温度下演化 10 ns的扩散径迹图 (a) T=100 K;(b) T=700 K;(c) T=1000 KFig.3.Diffusive trajectories of 1-SIA for temperatures of (a) 100 K,(b) 700 K and (c) 1000 K.
3.3 双自间隙原子扩散
图4给出了钨中2-SIAs的最近邻结构在不同温度下的扩散径迹.在达到转向温度1400 K之前,最近邻结构的扩散一直保持一维运动形式.当温度达到1400 K时,扩散由方向转向其他三个等价的方向,从而运动形式由一维转变为三维.随着温度的进一步升高,转向越频繁,但整个过程中一直保持最近邻状态,即使在高达2000 K的温度下也未解离.表1和表2的数据也表明了该结构极为稳定,其束缚能为 2.245 eV.

图4 最近邻结构在不同温度下演化 10 ns的扩散径迹图 (a) T=100 K;(b) T=1400 K;(c) T=2000 KFig.4.Diffusive trajectories of 2-SIAs-1st for temperatures of (a) 100 K,(b) 1400 K and (c) 2000 K.
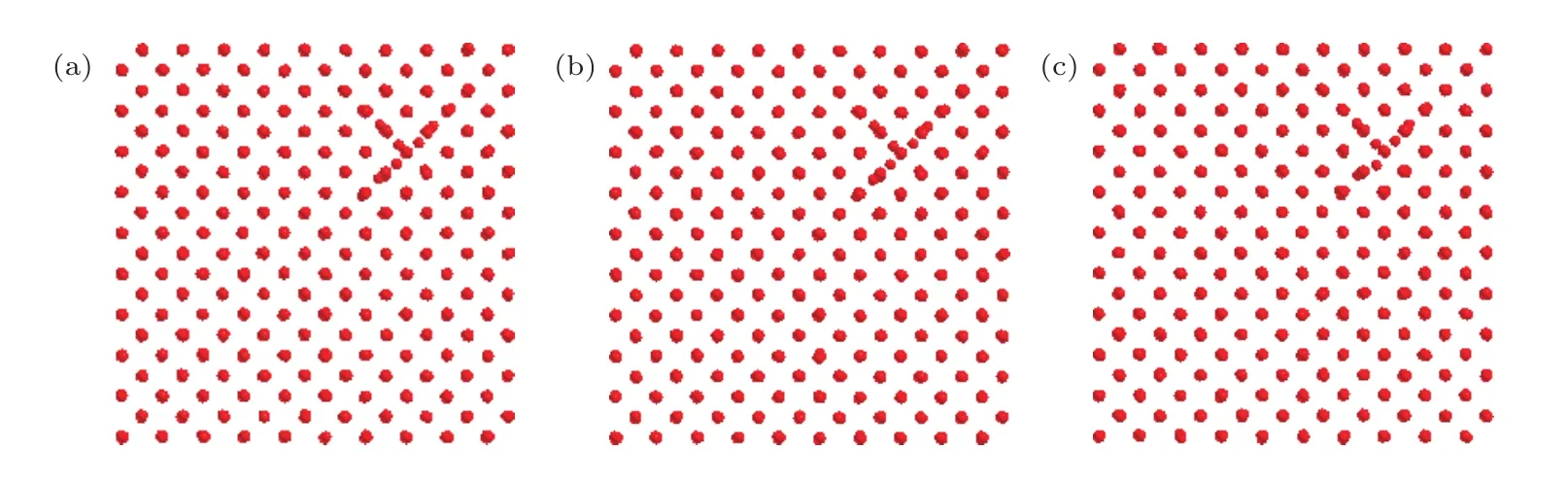
图5 T=500 K 时 sessile 结构在不同时间的结构图 (a) t=0.5 ns;(b) t=2 ns;(c) t=5 nsFig.5.Views of the sessile cluster obtained by molecular dynamics simulation at different time when T=500 K: (a) t=0.5 ns;(b) t=2 ns;(c) t=5 ns.
关于2-SIAs非平行的情况,主要研究了图2(d)所示的结构在不同温度下的动力学行为.在实际模拟时,将2-SIAs沿各自方向移动一定距离以便观察演化过程及结构变化.模拟表明,非平行的2-SIAs,沿各自方向扩散,一旦靠近,就会形成固着结构,具有相当的稳定性,演化径迹表明了这一过程.通过对其结构的分析,发现2-SIAs形成了如图5所示的sessile结构.sessile结构在一定温度范围内能长时间(ns)稳定存在.当温度达到1000 K时,sessile结构可以转化形成更为稳定的最近邻结构,随后按最近邻扩散方式进行扩散运动.前面束缚能的计算结果也表明最近邻结构较sessile结构更稳定.
SIAs团簇一直被认为具有高迁移率,在空间尺度上可以迁移相当大距离.直到Bacon等[24−27]的发现改变了人们的认识,即在某些情况下锆、钛、铁金属中SIAs团簇不再具有这种移动性,而是形成固着性的团簇.在材料受到辐照之后,在级联碰撞初期或热峰过程中SIAs相互作用都可能会形成sessile结构[28].但钨中sessile结构鲜见文献报道.本文研究结果表明,钨中也存在sessile结构,这有助于我们更全面地理解钨中的SIAs行为.如前所述,SIAs的动力学行为是决定材料微观演化过程的重要因素,这方面的精确知识是我们能正确理解和预测材料微观演化行为的必要条件.sessile结构在一定温度范围内不移动,与其他构型具有完全不同的行为属性.在动力学蒙特卡罗模拟中,如果单纯地考虑一种构型的SIAs,尤其是忽略掉sessile结构,很可能会导致模拟结果与实际情况出现大的偏差.该结构对材料微观演化行为的影响程度也是今后理论模拟和实验研究值得进一步探索的课题.
3.4 扩散系数
材料中缺陷的扩散行为会直接影响到缺陷的分布、生长演化等过程,从而对材料性能产生影响.扩散系数是衡量物质扩散能力的重要参数.为了进一步了解钨中SIAs不同构型的扩散特性,我们计算了它们在不同温度下的扩散系数.根据爱因斯坦公式[29],粒子的扩散系数可以表示为
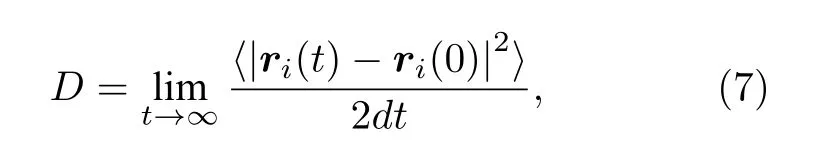
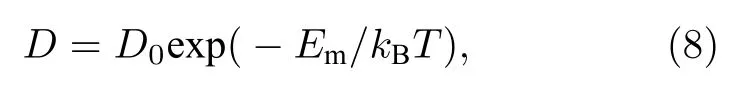
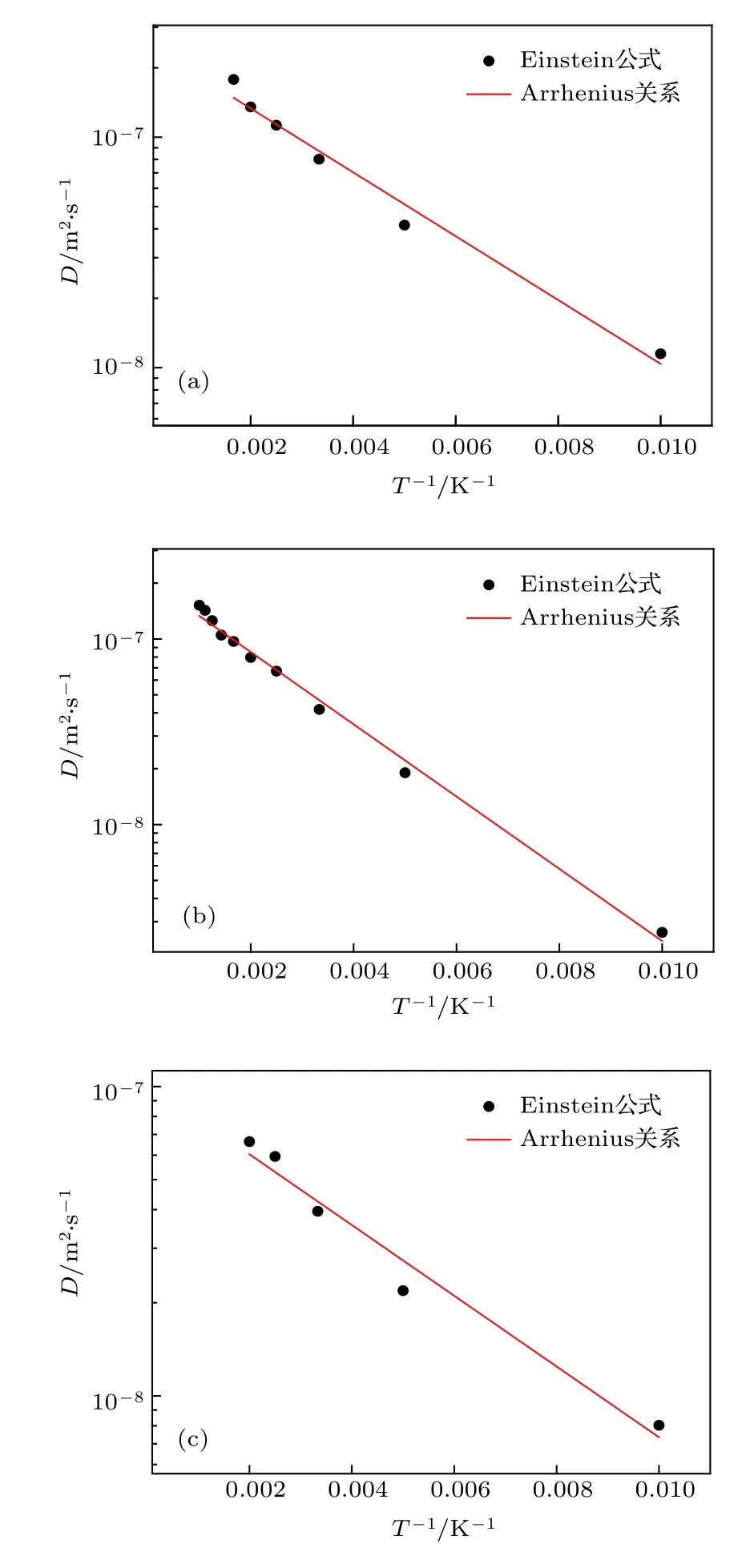
图6 不同结构的扩散系数 (图中实线是根据 Arrhenius关系拟合的结果) (a) 1-SIA;(b) 2-SIAs-1st;(c) 2-SIAs-2ndFig.6.Arrhenius plots of diffusion coefficients of single SIA and di-interstitial atoms in tungsten,which is determined using MD simulations and plotted as a function of the absolute temperature T: (a) 1-SIA;(b) 2-SIAs-1st;(c) 2-SIAs-2nd.

表3 Arrhenius拟合所得各个间隙结构的扩散迁移能与前因子Table 3.Migration energy Em (in eV) and prefactor D0 (in cm2/s) for W clusters diffusion obtained by Arrhenius fitting.
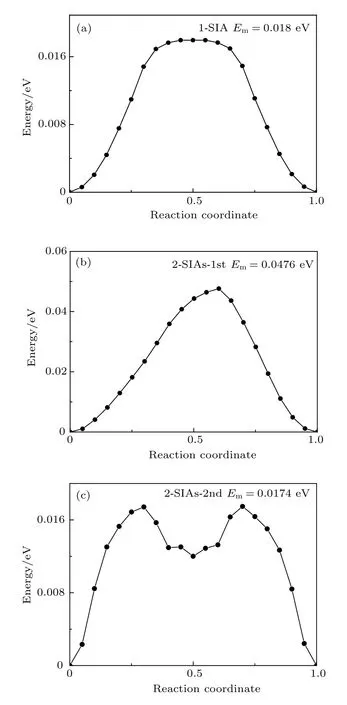
图7 通过 NEB 方法所得不同结构的迁移能垒 (a) 1-SIA;(b) 2-SIAs-1st;(c) 2-SIAs-2ndFig.7.Migration barriers for SIAs with different structures studied by NEB method: (a) 1-SIA;(b) 2-SIAs-1st;(c) 2-SIAs-2nd.
式中D0是扩散前因子,Em是迁移能,kB是玻尔兹曼常数,T是温度.随后将各种间隙结构的扩散系数进行Arrhenius拟合,获得了各个间隙结构的扩散迁移能.如表3所列,1-SIA的迁移能Em为0.0274 eV,D0为2.52×10–7m2·s–1;最近邻结构Em为 0.0386 eV,D0为 2.08×10–7m2·s–1;次近邻结构Em为 0.0248 eV,Do为1.02×10–7m2·s–1.
然而,最新的研究表明,对于单个钨 SIAs,其扩散规律并不满足Arrhenius关系,而是线性关系[30].这主要是因为,在所考察的温度范围下,其能量接近或高于钨SIAs的扩散迁移能.NEB方法通过搜索初末态之间的最小能量路径来确定迁移能垒,是最可靠的方法,能够准确地获得缺陷的扩散迁移能[31].为了确定上述几种结构的扩散规律能否用Arrhenius关系描述,采用NEB方法计算了这些间隙结构的迁移能.然后对比这两种方法获得的结果,若两者接近,则说明在目前的势函数下,扩散满足Arrhenius关系,否则就不满足.
图7给出了由NEB所获得的反应坐标与能量的关系.1-SIA,2-SIAs-1st和 2-SIAs-2nd 的迁移能 分 别 为 0.018,0.0476 和 0.0174 eV,这 与 由Arrhenius关系获得的结果(分别为0.0274,0.0386和 0.0248 eV)有明显差异,对比结果见表 3.这说明1-SIA和2-SIAs的扩散规律都不满足Arrhenius关系.于是对这些扩散数据进行线性拟合,拟合公式如下:
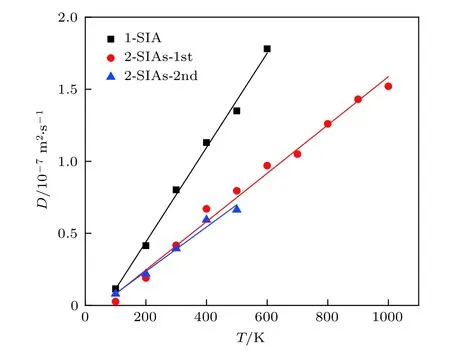
图8 不同缺陷的扩散系数Fig.8.Diffusion coefficient for self-interstitials of different configuration in tungsten determined by molecular dynamics simulations and plotted as a function of the absolute temperature T (the solid lines are linear fits).
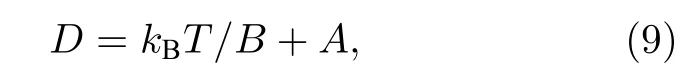
式中,kB是玻尔兹曼常数,kB=1.38 × 10–23 J·K–1;T是温度,单位是K;B和A为拟合常数.如图8所示,这些间隙结构的扩散系数与温度形成良好的线性关系.从图8可以看出2-SIAs结构的扩散能力明显低于1-SIA,在一定温度范围内2-SIAs-1st和2-SIAs-2nd的扩散能力接近.
4 结 论
由于SIAs及其团簇对材料微观性能的重要影响,本文运用分子动力学方法重点研究了钨中2-SIAs的不同构型在不同温度下的扩散行为,考察了它们的热稳定性和运动特性.结果表明: 2-SIAs不同构型的稳定性差异较大,扩散行为也有明显不同.彼此互为最近邻的双自隙原子结构最稳定,随着温度的升高,从一维扩散演变成三维扩散,在2000 K的温度下都没有出现解离;次近邻2-SIAs呈现出一维的扩散行为,在温度高于600 K将解离成两个独立运动的SIAs;而三近邻结构热稳定性最差,在温度高于 300 K 就将解离.钨中2-SIAs按非平行方式扩散会形成具有固着性的sessile结构,这一结构具有相当的稳定性,在一定温度范围内几乎不移动,随着温度升高将转变成最近邻结构,然后按最近邻结构的方式扩散运动.因此,在讨论2-SIAs团簇的动力学特性时不能同一而论,需考虑构型的影响.
最后,通过将NEB方法获得的SIAs迁移能与Arrhenius拟合的结果进行对比,表明了钨中1-SIAs和2-SIAs的扩散系数随温度变化的规律不满足Arrhenius关系,而是满足线性关系.这些结果给我们提供了新的视点,加深了我们对钨中SIAs的扩散行为的认识.
