诺氟沙星胶囊的质量分析与研究
2019-06-01李俊杨玉琴王英瑛周谊陈秀秀
李俊 杨玉琴 王英瑛 周谊 陈秀秀
(台州市食品药品检验研究院,台州 318000)
诺氟沙星(norfloracin)又名氟哌酸,为第三代喹诺酮类抗菌药,最早分别是1983年在意大利上市[1]和1984年在日本上市。诺氟沙星口服后部分吸收,血药浓度低,但尿、肠道浓度高,对革兰阴性菌和阳性菌引起无并发症的泌尿道感染疗效较好,临床上适用于敏感菌所致的泌尿道、肠道、耳喉鼻科、妇科、外科和皮肤感染性疾病[2]。国内诺氟沙星单方制剂一共有802个批准文号,其中诺氟沙星胶囊有697个的批准文号(规格均为0.1g),诺氟沙星片有52个批准文号。执行的法定标准为中国药典2015年版二部[3],国外英国药典(BP)、美国药典(USP)、日本橙皮书(JP)均没有收载诺氟沙星胶囊,收载有诺氟沙星片。诺氟沙星胶囊被列入2017年浙江省药品质量风险考核计划,共抽取样品57批次,抽样地点覆盖浙江省11个地市,而且分布均匀性较好;涉及生产企业20个和批准文号20个;抽自经营单位有45批,使用单位11批次,生产单位1批次。
对于诺氟沙星有关物质的控制,目前国外药典中JP16版[4]采用薄层色谱法,而EP9.3版[5]、BP2015版[6]和USP41版[7]方法均采用HPLC梯度洗脱法,虽然中国药典方法采用的也是HPLC梯度洗脱法,但从色谱条件、梯度程序、控制的杂质指标及限度等多方面均有很大的差异,而且国内也没有文献对现行有关物质方法进行研究的报道,因此有必要对国内外诺氟沙星的有关物质方法进行比较研究。关于溶出度方法,国外药典没有收载诺氟沙星胶囊,国内外药典方法的诺氟沙星片的溶出度方法比较接近,而国内现行2015年版中国药典诺氟沙星胶囊的溶出度方法与诺氟沙星片一致,但近几年来国内药品质量公告及文献[8]显示采用现行的溶出度方法诺氟沙星胶囊溶出度不合格的情况比较多,因此溶出度的研究也很有必要。
为了考察药品的质量状况以及有效性、安全性和质量可控性,本文根据现行法定标准的检验结果结合探索性研究,对市场上的诺氟沙星胶囊产品质量进行了分析和研究,并提出了相应的改进建议。
1 仪器与试药
Waters e2695高效液相色谱仪;岛津UV-2550紫外可见分光光度计,Agilent 708-DS溶出度仪(配备850-DS自动取样器),瑞士万通KF870水分滴定仪
来自国内20个生产厂家的57批诺氟沙星胶囊均为2017年浙江省药品质量风险考核抽验样品,规格均为0.1g;诺氟沙星原研片,日本杏林制药株式会社,规格100mg,批号:S001;诺氟沙星对照品(中国食品药品检定研究院,批号:130450-201206)、诺氟沙星杂质A(中国食品药品检定研究院,批号:130610-201502,99.7%)、杂质定位用诺氟沙星对照品(含杂质K) (EP标准品,批号:1.0),诺氟沙星系统适用性对照品(EP标准品,批号: 2.0);氢氧化钠、冰乙酸、氯化钠、盐酸、磷酸二氢钾、磷酸氢二钠、磷酸为分析纯,甲醇为色谱纯。
2 实验方法
2.1 法定检验方法
57批样品按照法定标准中国药典2015年版二部进行检验。
2.2 探索性研究
2.2.1 溶出度
对现行标准的溶出度测定方法的影响因素进行考察,具体考察溶出介质的不同pH值、不同离子强度及配制方法对溶出度结果的影响,不同pH计,不同溶出度仪等因素对溶出度测定结果的影响。
溶出曲线的考察采用日本杏林制药株式会社的诺氟沙星片为参比制剂,参考JP的溶出度条件,分别在pH1.2、4.0、6.8和水4种溶出介质条件下对9个生产厂家的10批样品及原研片进行溶出曲线的测定,考察诺氟沙星胶囊样品和原研片的溶出状况,并通过计算f2相似因子法评价溶出曲线的相似性。
2.2.2 水分
采用水分滴定法和烘干法测定样品的水分含量,并对水分对溶出度的影响进行考察和研究。
2.2.3 有关物质
参照EP 9.3版建立的有关物质测定方法,采用Supelcosil LC-ABZ色谱柱(4.6mm×250mm, 5μm),以磷酸溶液[取水用磷酸调节pH值至(2.0±0.1)]为流动相A,乙腈为流动相B;线性梯度洗脱(0~5min,B为5%;5~7min,B为5%→7%;7~10min,B为7%→13%;10~15min,B为13%→53%;15~20min,B为53%→90%),以265nm为检测波长,流速1.4mL/min,柱温60℃。
2.2.4 胶囊壳中铬的含量
采用原子吸收分光光度法对57批诺氟沙星胶囊抽样样品的胶囊壳中的铬含量进行了测定。
3 结果与讨论
3.1 法定检验结果
按照中国药典2015年版二部进行全检,结果57批样品合格率为91.2%。
不合格项目为溶出度,57批样品中有5批溶出度不合格,分别为5个不同的生产厂家,多批样品存在不同程度的个体差异较大的现象;装量差异结果显示,各生产企业之间、企业内部批次之间差异较大;有关物质结果显示:57批样品杂质A的含量均在0.1%以下,均符合标准规定的0.2%的要求,反映出杂质A的控制水平较好;对于其他单个最大杂质,除1批样品为0.49%,接近限度边缘,该企业应关注这个杂质,其余58批均在0~0.25%范围内,处于安全可控的范围内;有4批次样品的杂质总量相对较高,其余批次的杂质总量均在0.5%以下;含量测定结果均在90.0%~110.0%的范围内,其中1个厂家的3批样品含量在105%~108%之间,同时结合溶出度结果也相对偏高,提示该厂家应重视生产投料的准确性。
3.2 探索性研究结果
3.2.1 溶出度
对现行标准的溶出度方法的影响因素进行研究,结果显示:(1)溶出介质pH值和电解质强度对部分产品溶出度影响显著,pH值在规定值4.0附近波动0.1时,部分产品溶出度检查结果相差约8%,波动0.2时,溶出度检查结果相差约15%;pH计电极使用一段时间后,在正常情况下,旧的电极比新的电极测定值约大0.2。(2)按药典标准配制溶出介质,用老的电极测pH值结果约为标准规定的4.0,但用相对较新的电极测结果约为4.2,需加约1.2mL的冰醋酸调节pH值至4.0,而标准规定配制溶出介质时,加冰醋酸为2.86mL,是一个精确的量,本研究认为正常情况下再加这么多的冰醋酸不是“调节”,这可能与标准制订时没有考虑到pH电极新旧差异有关。(3)按中国药典2015年版二部测定部分批号的诺氟沙星胶囊样品时,不注意溶出介质的配制、pH计的性能以及测量的pH值不准确均会导致溶出度结果不正确甚至结论不对;对于按法定标准检验溶出度不合格的5批样品,采用不同溶出度仪、不同的实验人员进行进一步的分析和考察。结果发现,两批平均溶出度低于50%的样品,其不同仪器、不同的实验人员测得结论均不合格;但另有3批平均溶出度稍高但不合格的样品,其不同仪器、不同的实验人员测得结果有差异,结论不一致,详见表1。分析产生的原因除了胶囊样品个体之间的差异外,不同品牌溶出度仪在仪器各部件上的微小差异(如溶出杯底部的弧度、振动等)的综合而产生的系统误差可能也是导致上述情况的原因之一。由此可见,同一批样品使用经过校验均合格但品牌不同的溶出度仪结果差异较大,甚至影响到样品检测结果的判定。因此,本品溶出度结果在限度上下边缘时应考虑不同溶出度仪的影响。
对溶出曲线的研究结果显示,pH1.2的溶出介质条件下,各受试样品与参比制剂的溶出曲线相似,而pH4.0、水、pH6.8的溶出介质条件下,多批次样品的溶出曲线与参比制剂相比差异较大。
3.2.2 水分
57批样品的水分含量分布不均匀,在2.6%~16.7%的范围内分布。
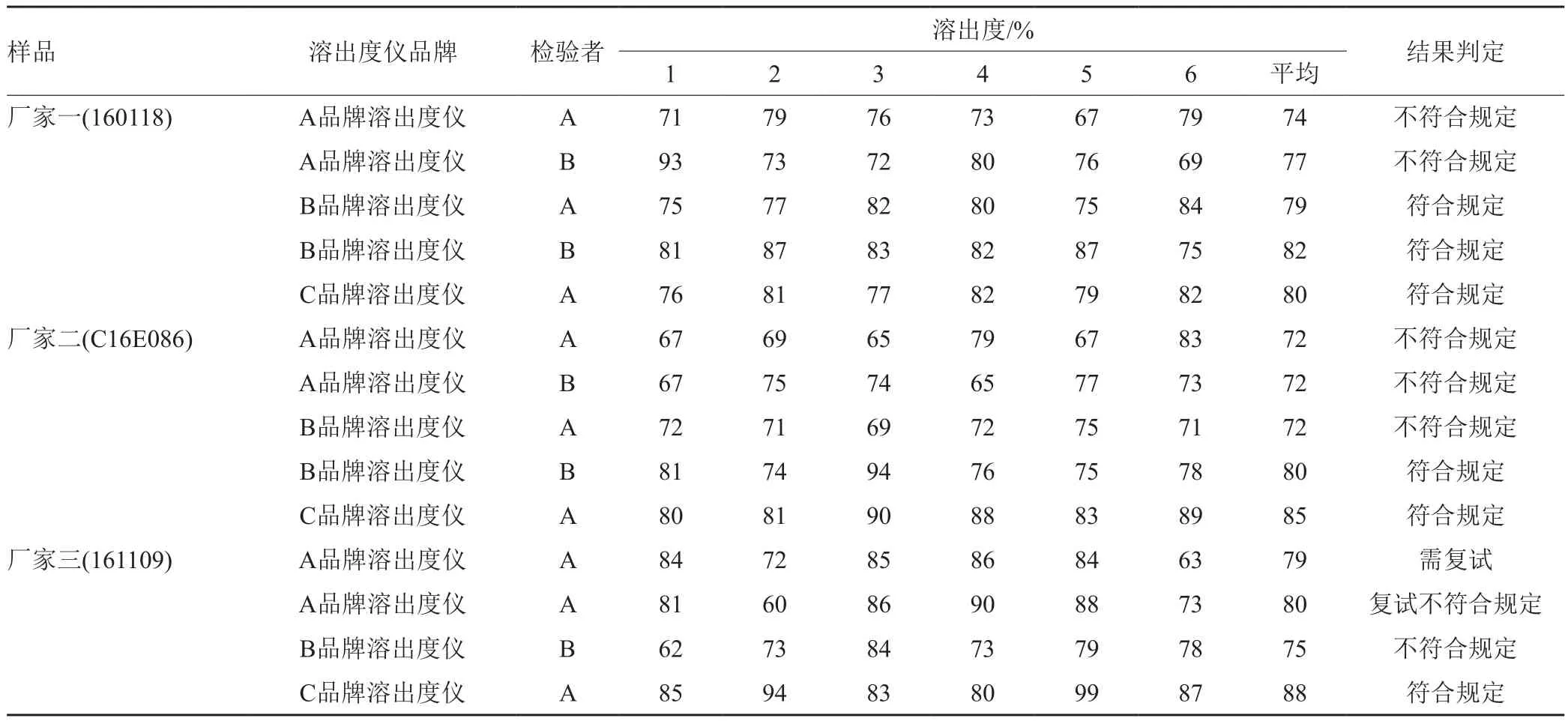
表1 不同品牌溶出度仪测得的部分样品的溶出度结果比较Tab.1 The comparison of results of dissolution measured by different brand dissolution apparatus
对57批样品的水分含量结果以及依标准测定的平均溶出度的结果,进行统计分析,结果显示:(1)随着样品中水分的增加,平均溶出度呈现下降的趋势,而从实际检验的结果看,样品测得的溶出度较高的样品其水分含量均相对较低,从统计学的意义上来说,诺氟沙星胶囊的溶出度与水分在一定程度上存在负相关;(2)57批样品中有15批在铝塑包装外增加了复合膜包装,这15批样品水分含量相对均较低,而平均溶出度结果均相对较高,说明样品的包装在一定程度上对水分有影响,进而会影响溶出度结果,文献[9]也显示增加背封袋可以减少样品在储存过程中溶出度不合格的问题;(3)某一生产厂家的7批次样品中有6批次采用复合膜包装的样品,其测得的平均溶出度结果均在90%以上。另有1批次生产日期稍早的样品没有加复合膜包装,其测得的平均溶出度结果较低,处于合格边缘,但其水分含量比其他6批样品并没有高出多少,相差不大,推断原因可能是在没有改变空心胶囊来源的情况下,不加复合膜包装可能对样品胶囊壳的性能有较大的影响,进而影响到样品溶出度的结果。根据文献[10],明胶胶囊在高湿度环境下会发生构象变化和交联,形成一个膨胀、橡胶状的、水不溶膜,阻滞或限制药物的释放。
3.2.3 有关物质
参照EP9.3版[5]、BP2015版[6]和USP41版[7]建立的有关物质测定方法对抽验的57批样品进行了有关物质测定,结果3个生产厂家的5批次样品中检出杂质E超标(限度为0.15%),1批检出杂质K在0.15%的限度边缘,结果见表2,色谱图见图1。
杂质E为诺氟沙星结构式中F与Cl发生竞争置换得到的副产物[11],而杂质K为诺氟沙星结构式中1号位上乙基改为甲基的化合物,这两个杂质均为诺氟沙星原料在合成过程中产生的工艺杂质,结构均较稳定,同时经过强降解实验研究也没有发现这两个杂质产生,因此诺氟沙星原料中杂质控制不严是诺氟沙星胶囊样品中杂质E超标和杂质K处于限度边缘的主要原因,同时诺氟沙星原料有关物质方法也没有对这两个杂质进行控制,无法检出杂质E,也成为杂质超限的另一个原因。
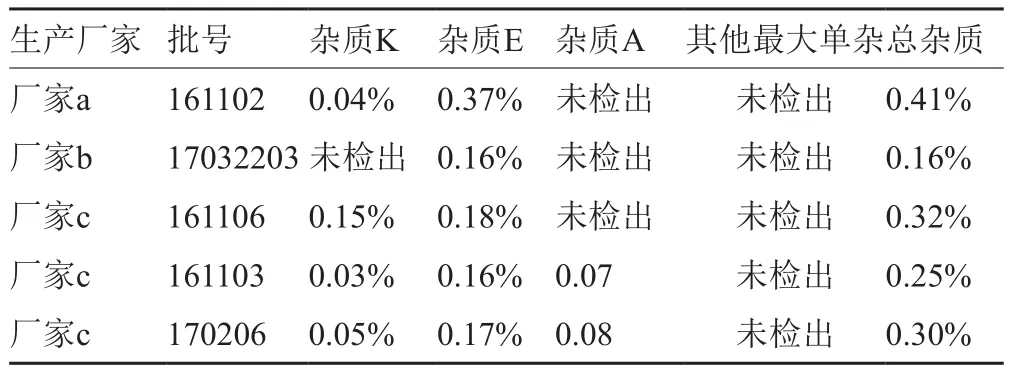
表2 部分有关物质超标的样品测定结果Tab.2 Results of related substances exceeding the standard
对法定标准方法和新建立的方法进行了比较研究,色谱图详见图2。结果发现:现行中国药典2015年版的诺氟沙星及诺氟沙星胶囊的有关物质测定方法已知杂质控制指标少,仅控制已知杂质A,没有控制杂质E、K等杂质,色谱条件也不合理,难于检出已知杂质E。分析其产生的原因:(1)色谱条件规定的初始流动相比例中有机相比例过高,梯度条件不适合,导致诺氟沙星主峰柱效较低,与相邻的杂质E峰难于分开。实验研究表明,在不改变流动相初始比例及梯度条件的情况下,采用不同品牌的C18色谱柱均无法很好地分离诺氟沙星主峰与杂质E,一些品牌色谱柱的色谱图中杂质E与诺氟沙星主峰完全重叠;(2)不同品牌的C18色谱柱对测定结果影响较大,实验研究表明,在改变和调整流动相初始比例及梯度条件的情况下采用3个不同品牌的C18色谱柱进行分离,仅有1个品牌的色谱柱可以使诺氟沙星主峰与杂质E得到很好地分离。
因此,诺氟沙星胶囊现行标准的有关物质方法不能有效控制药品的质量;诺氟沙星色谱峰与杂质E峰重叠出峰,也会导致含量测定方法专属性差,需改进提高。
3.2.4 胶囊壳中铬的测定
胶囊壳的铬含量的探索性研究结果显示,57批样品的胶囊壳的铬含量质量状况较好,安全风险低。
4 小结

图2 法定标准有关物质方法与改进的方法的色谱图比较Fig.2 Comparison of HPLC chromatograms between statutory standards method and improved method of related substance
通过本次质量风险考核结果显示,诺氟沙星胶囊的样品存在一定质量问题,中国药典2015年版二部诺氟沙星胶囊质量标准需要修订提高。依现行标准检验有2批不合格,3批次因不同溶出度仪检测结论不一致,无法判断;探索性研究发现现行质量标准“溶出度”方法中溶出介质配制的规定可能忽略了pH值波动、离子强度的显著性影响;多个厂家的样品的溶出曲线与原研制剂差异较大;样品的水分、包装均会影响溶出度的结果;5批样品有关物质检出杂质E超标,现行质量标准的“有关物质”方法中诺氟沙星色谱峰与杂质E峰重叠,难于分离,方法难于有效控制药品质量,须改进和提高。
