Mg在Si(220)表面吸附的第一性原理计算
2019-04-29张佳佳李晓琴
苏 蓉,李 娜,张佳佳,李晓琴,赵 辉
(天津师范大学 物理与材料科学学院,天津 300387)
几乎所有金属都能与Si表面发生反应生成硅化物,而金属/硅化物体系不仅被广泛应用于工业目的,同时具有重要的科研价值[1].其中,碱金属和碱土金属在硅表面的吸附引起了研究人员的特别关注,尤其是在Si表面吸附Mg原子[2].将Mg原子吸附到Si衬底上可以生长出Mg2Si薄膜.Mg2Si是一种重要的带隙半导体,在光电子领域具有潜在的应用价值.此外,Mg2Si是一种无毒、无污染的材料[3],其组成元素Mg和Si在地球上的含量丰富,资源寿命长,是一种环境友好型半导体.
实验研究方面,国内外研究人员采用不同的实验方法和手段将Mg原子沉积在Si表面上[4]制备Mg2Si薄膜.Hosono等[5]通过在Mg蒸汽下加热块状Si(111)获得了晶向为(211)的多晶Mg2Si薄膜.Mahan等[6]通过分子外延技术在200℃下将Mg沉积在Si(111)基底上获得了具有(111)晶向的多晶Mg2Si薄膜.Yang等[7]基于放电等离子烧结法利用Mg和Si粉末制备了Mg2Si块体.Wittmer等[8]利用脉冲激光退火沉积技术将Mg沉积在Si(111)衬底上制备得到Mg2Si薄膜.
目前,有关Mg在Si晶面吸附的理论研究相对较少,Ying等[9]利用第一性原理对不同覆盖度(1/4,1/2和1 ML)下Si(111)表面吸附Mg原子的过程进行研究,揭示了Mg原子在Si表面的吸附机制.本研究采用基于密度泛函理论(DFT)的第一性原理方法构建了Mg在Si(220)表面的吸附模型,通过计算对其结构的稳定性和吸附特性进行分析,以期为在Si(220)衬底上沉积Mg制备Mg2Si薄膜提供理论依据.
1 计算模型与方法
为了研究Mg原子与Si(220)表面的相互作用,采用基于密度泛函理论的第一性原理研究该体系的电子结构.采用Material studio 6.0软件包中的CASTEP(cambridge sequential total energy package)模块进行计算.CASTEP是基于密度泛函理论的平面波赝势方法的量子力学程序.计算中,选用广义梯度近似(GGA)下的PW91泛函描述电子间的交换相关能,采用Mokhorst-Pack方法对K点进行取样.平面波截止能设定为230 eV,不可约布里渊区的K点取样为3×3×1,体系中各原子核内层电子与外层电子间的库仑吸引势采用超软赝势(ultrasoft pseudo potential).为了获得稳定结构,采用BFGS方法对未计算模型进行几何优化.在进行自洽计算时,原子总能收敛值取为2×10-5eV/atom,设置平均原子力小于0.5 eV/nm,最大原子位移容差小于0.02 nm.Mg原子和Si原子的原子轨道分别为2p63s2和3s23p2.
为了测试赝势平面波计算的可靠性,对于原始Si晶胞,使用7×7×7的K点网格和450 eV的平面波截止能进行收敛测试.优化后的晶格参数a0=0.549 5 nm,弹性模量B0=87.22GPa.计算结果与实验值(a0=0.5430 nm,B0=98.80 GPa)吻合良好[10].在优化好的晶胞上建立Si(220)表面结构,再将Si(220)表面构建成2×2×1的超胞结构,纵向采用7个Si原子层,在表层上方加1.2 nm真空层,建立基底模型,如图1所示.图1中绿色原子表示顶层硅原子,蓝色原子表示其余6层的硅原子.然后再对该模型进行几何优化,优化时允许所有原子进行驰豫.
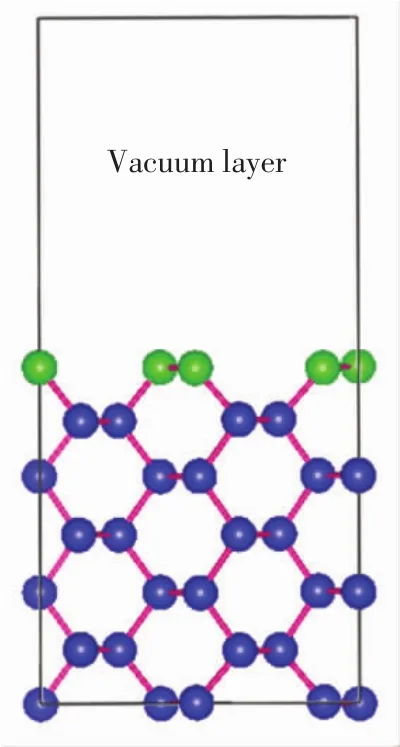
图1 构造的Si(220)超胞表面主视图Fig.1 Front view of the constructed Si(220)super cell surface
2 结果与讨论
2.1 吸附能
固定优化后Si(220)基底模型最下方的4层原子,允许上面3层原子运动.然后把Mg原子吸附在基底模型表面,Mg原子在Si(220)表面有顶位(T)、桥位(B)和穴位(H)3种可能的吸附位置,如图2所示.

图2 吸附位置的俯视图Fig.2 Vertical view of adsorption site
为了进一步确定Mg原子在Si(220)表面最稳定的吸附位置,本研究对Mg原子在Si(220)面上不同位置的吸附能进行计算,结果如表1所示.吸附能是把Mg原子从Si(220)表面分离所需要的能量,Ead=EMg/Si(220)–ESi(220)-EMg,其中EMg/Si(220)是吸附了Mg原子的Si(220)表面结构的总能,ESi(220)是未吸附Mg原子时Si(220)表面结构的能量,EMg为孤立Mg原子的能量.计算EMg的方法是在构造的边长1nm的立方原胞中放置1个Mg原子,然后通过几何优化求出.吸附能越低表明表面结构吸附原子的能力越强,最终获得的吸附结构越稳定[11].
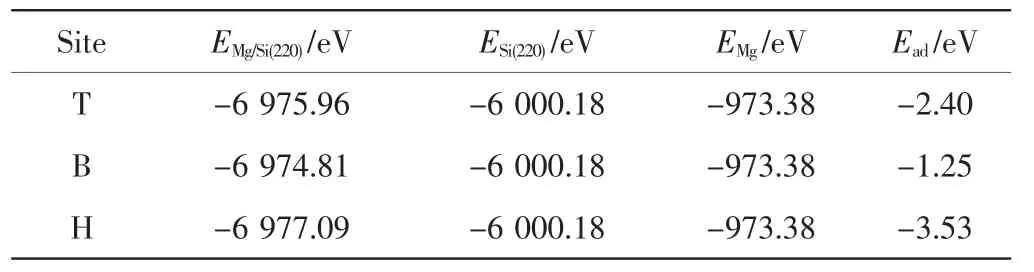
表1 Mg/Si表面体系优化后的能量Tab.1 Energies of Mg/Si surface system after optim ization
负值的吸附能表明吸附体系比未吸附体系更稳定,且数值越小,吸附的构型越稳定,所以通过比较吸附能的大小即可确定吸附的最优位置.由表1可知,Mg原子在Si(220)表面的顶位、桥位和穴位的吸附能均为负值,表明其吸附构型均可形成相对较稳定的结构.但对比吸附能的大小可知,Mg原子在穴位的吸附能小于桥位和顶位的吸附能,说明Mg原子在穴位比在桥位和顶位更容易被吸附[12].
2.2 吸附构型和功函数
Mg原子最初被放置在Si(220)表面顶位(T位)、桥位(B位)和穴位(H位)3个高对称吸附点.吸附结构经过弛豫达到最小的能量结构,通过计算吸附能已知Mg原子优选吸附在Si(220)表面的穴位.进一步计算3种吸附结构Si—Si键和Mg—Si键的键长、吸附表面Mg原子的垂直高度(dh)以及它们的功函数,相应的计算结果如表2所示.
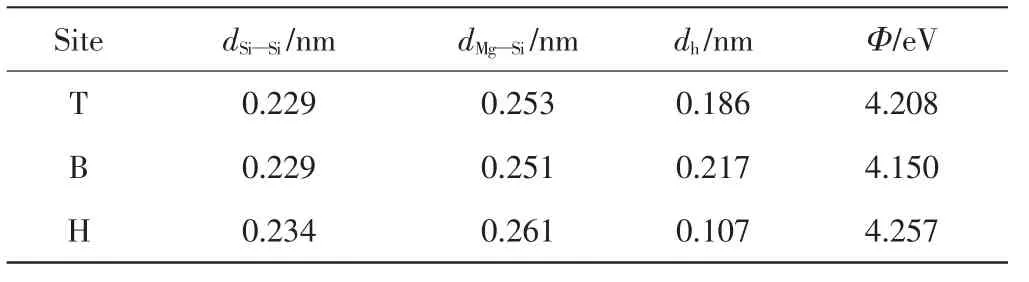
表2 键长dSi—Si和dMg—Si以及垂直距离dh和功函数ΦTab.2 Bond length dSi—Si and dMg—Si,the perpendicular distance dh and work functions Φ
由表2可知,通过比较吸附位点附近Si—Si键的键长,发现穴位(H位)的键长最长,且Mg原子与吸附位点相连的Mg—Si键的键长也是穴位(H位)的最长,说明穴位(H位)处的吸附形变最大.对于穴位吸附,吸附表面的Mg原子的垂直高度(dh)最低,说明此位点比顶位和桥位具有更强的吸附力.因此,Mg原子在Si(220)表面会优先吸附在穴位上.
半导体功函数是指真空中静止电子与半导体费米能级的能量差.电荷发生转移时,半导体的功函数发生变化[13].未吸附Mg原子的Si(220)表面的功函数为4.643 eV.由表2可知,顶位、桥位和穴位的功函数均小于理想Si(220)表面的功函数.功函数变小说明有电荷从Mg转移到Si(220)表面.
2.3 电荷布居
通过电荷布居分析可以定量讨论电荷的转移情况,本研究对以上3种吸附结构的Mg原子与最邻近Si原子的电荷布居进行分析,结果如表3所示.
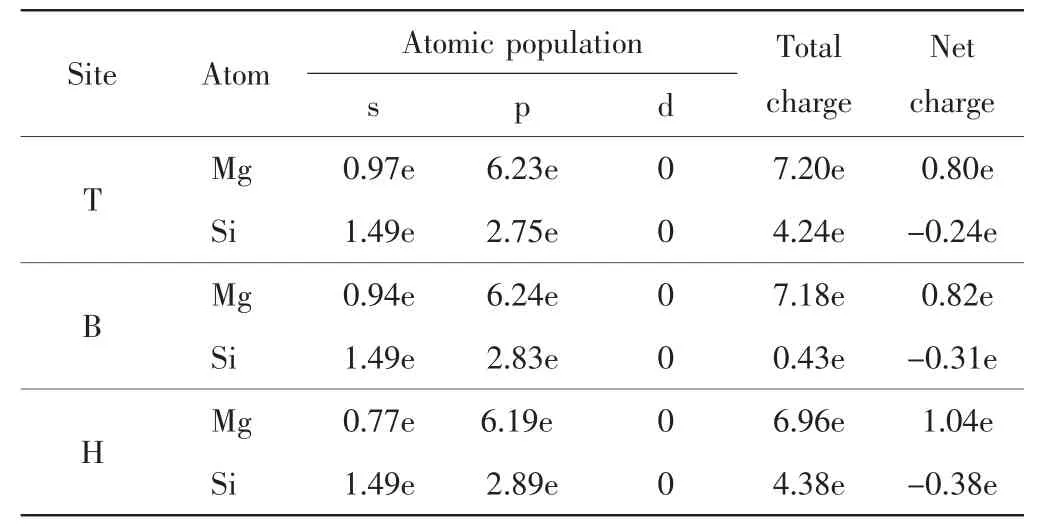
表3 不同吸附位置Mg原子与最近Si原子的电荷布居数Tab.3 Net M ulliken charges of Mg atom on the nearest Si at different adsorption sites
Mg原子失去电子表现为金属性,Si原子获得电子表现为非金属性.不同吸附位置Mg和Si原子间的得失电子情况不同.顶位、桥位和穴位Mg原子失去电子数分别为0.80、0.82和1.04,说明Mg原子在相对稳定的穴位处失去电子的能力更强,失去的电子数最多[14].由此可知,与吸附在顶位和桥位相比,Mg原子吸附在Si(220)晶面的穴位时,Mg和Si原子间存在较强的离子键和共价键,Mg与Si之间的作用力更强,形成的吸附结构也更稳定.
2.4 Mg/Si(220)吸附体系的电子态密度
电子态密度图反映了吸附原子与吸附晶面间电子态的分布和相互作用情况.本研究对Mg原子吸附于Si(220)表面穴位的情况进行分析,得到Mg原子在Si(220)表面穴位吸附前后体系的分波态密度(PDOS)和总态密度(TDOS),结果如图3所示.
图3(a)和图3(b)分别为Mg原子吸附前后的分波态密度(PDOS).由图3(a)可以看出,费米能级附近存在电子分布,表现出很强的金属性.由图3(b)可以看出,费米能级附近几乎无电子分布,表现出半导体的性质[15].比较图3(a)和图3(b)可知,Mg原子吸附在Si(220)面后,Mg原子的2p电子从0 eV附近移动到-10 eV附近,能量有所降低,且Mg原子3s电子的峰值降低明显.
图3(c)和图3(d)分别是吸附前后Si(220)面的分波态密度(PDOS).对比图3(c)和图3(d)可知,在吸附后的-7.5~-5 eV能量区间内,Si原子的3p电子向右移动,能量比吸附前有所增加.在0~2.5 eV能量区间内,吸附后Si原子3p电子的峰值不但有所降低,且峰的数量减少1个.对于Si(220)面来说,吸附前后变化明显的是Si原子的3p电子.
图3(e)和图3(f)分别为吸附前后体系的总态密度(TDOS).对比图3(e)和图3(f)可知,吸附Mg原子前,Si(220)面的价带在-12.5~0 eV能量区间,导带在0~3 eV能量区间;吸附Mg原子后,Si(220)面的价带有-45~-43 eV和-12.5~0 eV共2个能量区间,导带在0~2.5 eV能量区间.对比图3(c)和图3(e)可知,价带能量区间和导带能量区间均主要由Si 3s和Si 3p电子贡献,但最主要贡献来自于3p电子,3s电子的贡献相对较小[16].此外,吸附Mg原子前,-45~-43 eV能量区间内未出现峰值,吸附后却出现了1个小尖峰,这表明Mg原子吸附在Si(220)面后,电荷间的相互作用导致了该尖峰的出现.
综上所述,Mg原子吸附在Si(220)表面后,总态密度(TDOS)在较低能量区间-45~-43 eV内出现了一个较为明显的峰值,这个峰由Mg原子与吸附面的Si原子相互作用产生[17],因此,Mg原子和Si表面的相互作用主要源于Mg原子的3s、2p电子和Si原子的3s、3p电子.
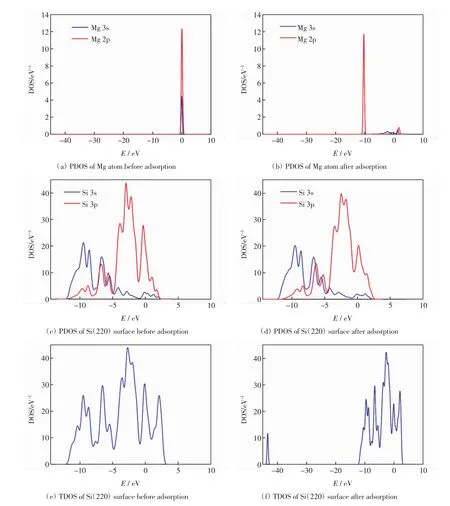
图3 Mg原子和Si(220)表面穴位吸附前后的态密度(DOS)Fig.3 DOS of Mg atom and Si(220)surface before and after adsorption on hollow site
3 结论
本研究采用第一性原理方法对Mg原子在Si(220)晶面的表面吸附行为进行计算,研究结果表明:
(1)吸附的最稳定结构是Mg原子的初始位置在Si(220)表面的穴位.此时的吸附能为-3.53 eV,属于稳定吸附.
(2)当Mg原子在Si(220)表面的吸附处于穴位时,吸附形变最大,且被吸附Mg原子与吸附面的距离最近.这说明此位点比顶位和桥位具有更强的吸附力,吸附结构更稳定.
(3)Mg原子吸附在Si(220)表面后功函数均变小,说明Si(220)表面吸附Mg原子后电子发生转移.
(4)Mg原子吸附在Si(220)表面的穴位时,Mg与Si原子间存在较强的离子键和共价键,增强了Mg与Si间的作用力,形成了更稳定的吸附结构.
(5)Mg原子吸附在Si(220)表面后,总态密度(TDOS)在较低能量区间内出现1个较为明显的峰值.该峰由Mg原子的3s、2p电子和Si原子的3s、3p电子相互作用产生.
