紫草素钠脂质体的制备及性能研究
2019-01-24施磊,叶隽,宋磊
施 磊,叶 隽,宋 磊
(1.上海市静安区食品药品检验所,上海 200435; 2.上海市交通大学医学院附属同仁医院,上海 200050)
紫草有消炎[1]、抗菌[2]、抗肿瘤[3]、抗病毒[4]等多种药理作用,多用于治疗褥疮、尿布疹、皮肤烧伤、湿疹等各种皮肤炎症,临床应用前景广阔[5-6]。临床研究发现紫草中的主要活性物质紫草素(shikonin)不仅具有抗菌消炎和抗氧化等多种药理作用,而且能够促进皮肤生长和伤口愈合[7]。由于紫草素为脂溶性活性物质,因此目前常见的紫草制剂多为油性基质的膏剂,活性成分易氧化变质,且吸收性和舒适度较差。此外,近年来研究发现紫草科植物中多含有吡咯里西啶类生物碱(pyrrolizidine alkaloids,PAs),这是一种肝毒性植物毒素,长期或过量应用会损害肝脏[8-10]。因此,欧美国家紫草相关制剂的应用必须严格按照处方或医嘱。而我国目前市场上的大部分紫草相关制剂依旧直接以紫草提取物入药,为临床应用带来极大的安全隐患。
针对这些不足,近年来关于紫草素新型制剂的开发研究已有报道[11-12],但这些研究均是以紫草提取物为主药,其成分复杂,含有较多杂质,不仅影响药效,而且PAs的潜在危害依然存在。因此,基于这一现状,本研究拟将紫草提取物提纯,并将其以碱性有机盐的形式制备成脂质体,对其形貌、包封率和体外释放等性质进行考察,为安全的新型紫草制剂的开发提供参考。
1 实验仪器及材料
1.1 仪器
沃特世E-2695液相色谱仪(沃特世科技有限公司),Hitachi H7650透射电子显微镜(HITACHI公司),PHS-3C型pH计(上海雷磁科学仪器股份有限公司),JY92-IIN超声波细胞粉碎机(宁波新芝生物科技股份有限公司),ZS90纳米粒径电位分析仪(英国马尔文公司)。
1.2 材料
左旋紫草素对照品(批号,110769-200506,成都普思生物科技有限公司),紫草(上海市童涵春堂中药店),甲醇(色谱纯,德国Merck公司),其他试剂均为分析纯。
2 方法与结果
2.1 紫草素的提取和提纯
取5 g紫草粉末浸没于60 ml 石油醚中, 65 ℃搅拌3 h,冷却后过滤,加入新的石油醚60 ml重复提取2次。合并滤液,浓缩得紫草提取物粗品0.71 g,提取率为14.2%。将该粗品溶于60 ml石油醚中并加入等体积饱和碳酸钠溶液,40 ℃条件下剧烈搅拌4 h后,分离上层有机液,加饱和碳酸钠溶液萃取多次,直到下层不显蓝色,合并碱性水溶液,酸化后以乙酸乙酯萃取多次,直至上层无明显红色,合并有机层浓缩,得到纯化的紫草素0.14 g提取率为2.8%(以紫草粉末质量计)。
以HPLC检测紫草素提取物和提纯后的紫草素,检测结果见图1:以左旋紫草素对照品为参照,由图1C可知其出峰时间约为7.9 min,而紫草素提取物(图1A)和碱化提纯的紫草素(图1B)也均在7.9 min左右出峰,可见二者主要成分为左旋紫草素。但将紫草素提取物与后者相对比,发现紫草提取物中含有更多杂峰,这是由多种杂质引起的,而紫草素碱化提纯后则非常纯净,其纯度达到99.35%,除左旋紫草素之外,几乎不含有其他成分。
2.2 脂质体的制备
2.2.1紫草素脂质体
精密称取10 mg胆固醇,50 mg卵磷脂和5 mg紫草素粗提物溶于10 ml无水乙醇中超声溶解,并将其用注射器匀速注入已水浴恒温50 ℃ 的PBS溶液(pH 7.4,10 ml)中,反应30 min后取出,将溶液置于旋转蒸发器上35 ℃温度下减压蒸发,除去有机溶剂。剩余溶液于50 ℃保温1 h, 冷却后超声10 min,分别以0.45、0.22 μm的微孔滤膜过滤,得到碱化紫草素脂质体,封装并于4 ℃保存。用同样方法制备空白脂质体。

图1 紫草提取物(A)、碱化紫草素(B)和对照品(C)的HPLC图 A.紫草提取物;B.碱化紫草素;C.对照品;1.左旋紫草素
2.2.2紫草素钠脂质体
为提高药物的体外释放度,先将紫草素溶于少量饱和碳酸钠中使之完全生成水溶性的紫草素钠盐,并以pH值为7.4的磷酸盐缓冲溶液定容至10 ml。其他物料的加入以及制备方法与紫草素脂质体制备方法完全相同。
2.3 脂质体的外观形态、粒径和Zeta电位的测定
将脂质体加适量超纯水稀释后制样,在透射电子显微镜下观察其外观形态,并用纳米粒径电位分析仪测其粒径大小和Zeta电位,结果如图2所示:粒径分析结果显示空白脂质体的平均粒径为98.11 nm,聚合物分散性指数(PDI)为0.117;而紫草素钠脂质体的平均粒径为104.2 nm,PDI为0.103。两种脂质体粒径大小均一,有明显的双分子层结构,均呈正态分布,且紫草素钠脂质体PDI值较小,说明紫草素钠脂质体的分散度较好。由Zeta电位图可以看出,空白脂质体的Zeta电位为-15.0 mV,紫草素钠脂质体的Zeta电位为-14.8 mV,说明以上两种脂质体的稳定性相当。将纯化的紫草素以钠盐的形式制备成紫草素钠脂质体,与空白脂质体相比粒径有所增加,且中心不透明,这是由于紫草素碱化成盐后亲水性增强,紫草素钠脂质体被包封在内水相中引起的。

图2 空白脂质体(A、B)和紫草素钠脂质体(C、D)的透射电镜图和粒径分布图
2.4 左旋紫草素含量测定[13-14]
色谱条件:色谱柱:Inertisl®ODS-3(4.6 mm×250 mm,5 μm);流动相:甲醇-0.025 mol/L磷酸(85∶15);流速:1.0 ml/min 检测波长:516 nm;柱温:25 ℃;进样量:20 μl。
标准曲线绘制:精密量取浓度为100 μg/ml的储备液0.50、1.00、2.00、3.00、4.00、5.00 ml,分别于10 ml的容量瓶中用甲醇定容,配制成5、10、20、30、40、50 μg/ml的对照品溶液,高效液相色谱检测并记录峰面积A,绘制峰面积A-对照品质量浓度C的标准曲线并进行线性回归分析,得到紫草标准曲线方程:A=3 429.474 2C-319.393 6(r=0.999 96)。结果表明紫草素在5~50 μg/ml范围内线性关系良好。
2.5 包封率测定
包封率是指包封在脂质体内的药物量占投料量的百分比,是评定脂质体制剂质量的一个重要指标。将脂质体溶于pH7.4的磷酸盐缓冲溶液中并定容至10 ml,量取3 ml加适量甲醇超声破乳,再加甲醇定容至5 ml,并在12 000 r/min条件下离心10 min,取上清液,测其浓度为脂质体中紫草素总量,记为C总(μg/ml);另取3 ml的紫草素钠脂质体混悬液在5 500 r/min条件下离心10 min,然后取上清液,测得未包入脂质体的游离紫草素浓度C游(μg/ml),然后根据公式(1)计算其包封率。
包封率=(C总-C游)/C总100%
(1)
实验表明,紫草提取物脂质体的包封率为20.4%(20.11%、20.42%、20.57%;RSD为1.2%),而紫草素钠脂质体的包封率为43.7%(44.02%、43.16%、43.84%;RSD为1.0%),与前者相比,紫草素碱化成盐后包封率提高1倍。其原因主要是由于包封率测试是以左旋紫草素为参照,在药物加入量相同的情况下,紫草素钠脂质体中紫草素纯度更高,因此表现出较高的包封率。
2.6 体外释药特性
采用动态透析法测定脂质体的体外释放特性:将脂质体溶于pH7.4的磷酸盐缓冲溶液中并定容至10 ml,并置于透析袋中,以含有0.5%吐温-80的磷酸盐缓冲溶液(pH7.4)为释放介质,(37±0.5)℃下搅拌,分别于1/3、2/3、1、2、4、6、8、10、12、24 h取样1 ml,同时补加相同温度相同体积的释放介质,测样品中紫草素浓度,并按公式(2)计算药物累计释放量Q。
(2)
式中:Q为药物累计释放量,单位为%;Cn为第n次取样的紫草素浓度,单位为μg/ml;Ve为取样体积,单位为 ml;Md为脂质体中包载的药物质量,单位为mg。
结果如图3所示,12 h内紫草提取物脂质体及紫草素钠脂质体的药物累计释放量分别为48.0%(47.53%,48.12%,48.21%;RSD为0.8%)和65.8%(65.97%,66.12%,65.36%;RSD为0.6%),这是因为紫草素碱化成盐后水溶性更好,与脂溶性紫草素脂质体相比,在磷酸盐缓冲溶液中释放更为彻底。

图3 紫草素脂质体(A)和紫草素钠脂质体(B)的药物累计释放曲线
3 讨论
本研究首先对紫草素提取物进行碱化提纯。紫草素是紫红色的蒽醌类脂溶性生物活性物质,因为具有酚羟基而显酸性,其侧链上的酯键在碱性条件下会水解,并与碱离子形成水溶性的紫草素盐,其颜色为蓝色,加入酸后,又可形成脂溶性的紫红色紫草素,其转化过程见图4。
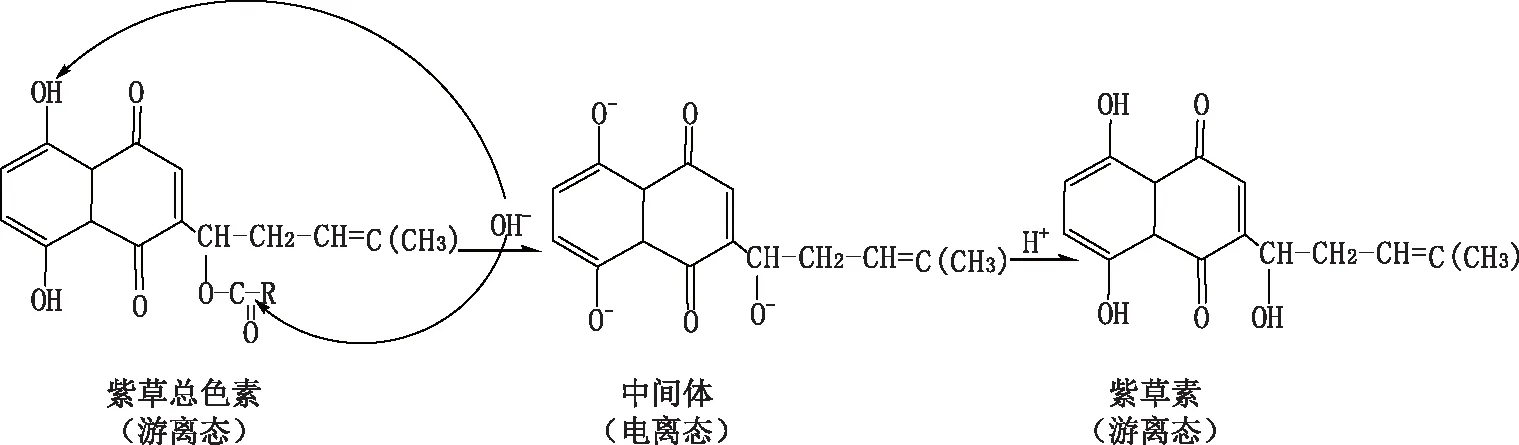
图4 紫草素碱化过程
经过碱溶酸沉法提纯后,紫草素被有效纯化,紫草提取物中大部分有害杂质均被除去(图1),其安全性大大提高,对解决现有市场上紫草相关制剂安全问题有积极意义。此外,对紫草素钠脂质体性质研究发现,与紫草素脂质体相比,由于所含药物纯度和亲水性更高,其脂质体包封率和体外释放度均比后者有较大提高,有利于提高药效,为开发安全、高效的紫草新型制剂提供思路。
