柱前衍生结合高效液相色谱-荧光法测定水中草铵膦
2018-07-31王姗姗俞瑞鲜王菲迪吴声敢赵学平
王姗姗,俞瑞鲜,王菲迪,吕 露,吴声敢,范 艳,赵学平
(浙江省植物有害生物防控重点实验室—省部共建国家重点实验室培育基地 农业部农药残留检测重点实验室浙江省农业科学院农产品质量标准研究所,浙江 杭州 310021)
草铵膦是一种膦酸类、广谱、非选择性、触杀性和非残留性除草剂。与草甘膦相比,草铵膦更能有效去除多年生恶性杂草[1-3]。尽管草铵膦属于非持久性农药,但其大量使用仍造成了严重的环境污染。大量研究表明,低剂量草铵膦会影响初生动物脑部发育,动物食用过量草铵膦会导致发抖、抽搐、呼吸困难等中毒症状。同时,草铵膦的不合理使用还会对水体造成污染,尤其对饮用水的污染将会对人体健康产生重大威胁[4-7]。因此,很有必要建立一种简便、快速的草铵膦检测方法。
草铵膦在结构中含有膦酸基、氨基、羟基,是极强的两性化合物。使用气相色谱和气相色谱-质谱联用技术进行草铵膦检测时,需要将其转化成可气化的物质,该操作会引入过多的其他试剂,且操作过程繁琐,检测效率和灵敏度都相对较低[8-11]。草铵膦紫外吸收弱,且很难在常规的C18柱子长保留,所以使用液相色谱-紫外测定时灵敏度较低[12-13]。使用液相色谱-质谱直接测定草铵膦时,仪器响应较低,且需要离子交换柱等特殊柱子进行分离。所以,目前文献报道中常采用氯甲酸-9-芴基甲酯对草铵膦进行柱前衍生,再结合液相色谱质谱法和液相色谱-荧光法进行草铵膦检测[14-19]。虽然固相萃取小柱可以去除反应体系中的硼酸钠盐和部分杂质,但仍不能完全去除衍生化试剂水解产物和一些影响草铵膦定量的干扰杂质。另外,衍生化试剂水解产物浓度高,进入质谱会造成离子源的污染,影响质谱的稳定性,干扰草铵膦的准确定量。本文采用液相色谱-荧光法在Waters Atlantis®T3色谱柱上对草铵膦衍生化产物进行分离,在不使用固相萃取小柱的前提条件下,排除了杂质对草铵膦定量的干扰,为水中草铵膦的检测提供了一个简便、快速、准确的方法。
1 材料与方法
1.1 材料与试剂
乙腈(色谱纯,Merck公司),乙酸铵(色谱纯,美国Tedia公司),饮用纯净水(杭州娃哈哈集团有限公司),丙酮(分析纯,上海凌峰化学试剂有限公司),二氯甲烷(分析纯,杭州双林化工试剂厂),四硼酸钠(分析纯,上海凌峰化学试剂有限公司),氯甲酸-9-芴基甲酯(分析纯,上海安谱实验科技股份有限公司),草铵膦(纯度97.5%,购自德国Dr. Ehrenstorfer GmbH)。
1.2 仪器与设备
Agilent Technologies 1260高效液相色谱仪(美国安捷伦公司,配有自动进样器、荧光检测器,紫外检测器),Waters Atlantis®T3液相色谱柱(250 mm×4.6 mm×5 μm,美国Waters公司),GEMINI C18液相色谱柱(250 mm×4.6 mm×5 μm,美国Phenomenex公司),CNWBON LC-C18固相萃取柱(500 mg/6 mL,上海安谱公司),OASIS HLB固相萃取小柱(500 mg/6 mL,美国waters公司),AB104-S电子天平(感量0.000 1 g,梅特勒-托利多国际贸易上海有限公司),OHAUS SPS202F 型电子天平(感量0.01 g,奥豪斯仪器常州有限公司),MX-S可调式混匀仪(北京大龙兴创实验仪器有限公司),HH-4 恒温水浴锅(江苏金坛市江南仪器厂),10 mL棕色样品瓶,移液枪,容量瓶等。
1.3 标准溶液、缓冲盐溶液和衍生化试剂的配制
草铵膦标准储备液和标准工作液。电子天平称取草铵膦标准品0.020 5 g(纯度为97.5%),用饮用纯净水定容至10.00 mL,得到浓度为2.00×103mg·L-1标准储备液。用饮用纯净水逐级稀释草铵膦标准储备液,得到浓度为0.200、0.100、0.050 0、0.020 0、0.010 0、0.005 00和0.002 00 mg·L-1的草铵膦标准工作液。
四硼酸钠缓冲溶液。称取3.80 g四硼酸钠,用饮用纯净水定容至100.0 mL,得到浓度为0.100 mol·L-1的四硼酸钠水溶液。
氯甲酸-9-芴基甲酯丙酮溶液:称取0.10 g氯甲酸-9-芴基甲酯,用分析纯丙酮定容至100.0 mL,得到浓度为1.0 mg·mL-1的氯甲酸-9-芴基甲酯丙酮溶液。
1.4 色谱条件
Waters Atlantis®T3液相色谱柱(250 mm×4.6 mm×5 μm),柱温40 ℃;进样量20.0 μL,流速。0.8 mL·min-1,流动相A为5 mmol·L-1乙酸铵水溶液(称取0.385 4 g乙酸铵,用饮用纯净水定容至1 L,0.22 μm滤膜过滤后使用),流动相B为色谱纯乙腈。梯度洗脱程序:0~1 min,90%A;1~12 min,90%A~50%A;12~13 min,50%A;13~14 min,50%A~10%A;14~17 min,10%A;17~17.1 min,10%A~90%A;17.1~25 min,90%A。
1.5 样品衍生
移取1 mL水样于10 mL棕色玻璃样品瓶中,分别依次加入0.5 mL 0.1 mol·L-1的四硼酸钠水溶液和1 mL 1 mg·mL-1的氯甲酸-9-芴基甲酯丙酮溶液,盖紧瓶盖,涡旋约30 s,在40 ℃的水浴下反应30 min。反应完成后,将全部液体转移至10 mL的刻度管中,加入4 mL的分析纯二氯甲烷,振荡1 min,室温下静置约10 min后取上层水样过0.22 μm滤膜,进行高效液相色谱检测。
2 结果与分析
2.1 衍生化条件的优化
通过对比不同的衍生化试剂体积(0.5、1.0和2.0 mL)、硼酸钠缓冲盐体积(0.5、1.0和2.0 mL)以及衍生化时间(0.5、1.0、1.5和2.0 h)来确定最优的衍生化条件。根据试验结果,最终选择衍生化试剂体积1 mL,硼酸钠体积0.5 mL和衍生化0.5 h作为草铵膦衍生化反应的最终条件。
2.2 固相萃取条件的选择
根据文献报道[15,18],比较CNWBON LC-C18固相萃取柱(500 mg/6 mL)和OASIS HLB固相萃取小柱(500 mg/6 mL)的净化效果表明,以上2种固相萃取小柱都无法去除衍生化试剂的水解产物和干扰草铵膦衍生化产物定量的杂质,故本实验中不再使用固相萃取小柱,而是选择合适的液相柱子排除杂质对草铵膦定量的干扰。
2.3 流动相和色谱柱的选择
根据文献报道[14],选择乙腈-5 mmol·L-1乙酸铵水溶液作为衍生化产物的色谱分离的流动相,通过调整两者的比例确定最优的液相条件。对Waters Atlantis®T3色谱柱(250 mm×4.6 mm×5 μm)和Phenomenex GEMINI C18色谱柱(250 mm×4.6 mm×5 μm)的分离效果考察结果表明,以上2种柱子都可将衍生化试剂水解产物和草铵膦衍生化产物很好的分离,但使用Phenomenex GEMINI C18液相色谱柱时,无法将草铵膦衍生化产物与其极性相似的干扰杂质分离,导致无法准确定量草铵膦(图1)。使用Waters Atlantis®T3色谱柱,采用1.4色谱条件进行草铵膦衍生物分离时,不仅可获得很好的峰形,而且可实现草铵膦衍生化产物与其干扰杂质的基线分离,从而保证草铵膦的准确定量(图2)。

图1 草铵膦衍生化产物(0.00 500 mg·L-1)的Phenomenex GEMINI C18柱色谱图

图2 草铵膦衍生化产物(0.00 500 mg·L-1)的Waters Atlantis® T3柱色谱图
2.4 线性关系、回收率和精密度
如图3所示,将0.002、0.005、0.010 0、0.020 0、0.050 0、0.100和0.200 mg·L-1的草铵膦标准工作液衍生化产物进样分析,以草铵膦浓度、草铵膦衍生化产物峰面积绘制标准曲线,得到回归方程为y=2 701.5x+1.305 2(R2=0.999 9)。

图3 草铵膦标准曲线
在自来水中添加3档浓度的草铵膦标准溶液,其浓度分别为0.003 00、0.300和30.0 mg·kg-1,每档浓度设5个平行样品。平均回收率和相对标准偏差见表1,该方法的检出限(S/N=3)为0.000 370 mg·kg-1,定量限(S/N=10)为0.001 23 mg·kg-1。
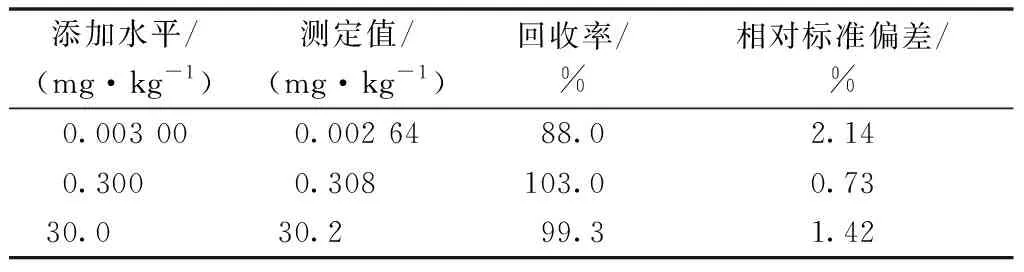
表1 不同添加水平下自来水中草铵膦的 回收率和相对标准偏差
3 小结
本文采用柱前衍生-高效液相色谱-荧光法建立了水中草铵膦的检测方法,该方法不需要固相萃取小柱即可排除杂质对草铵膦定量的干扰,且由于排除了杂质的干扰,使得该方法的检出限为0.000 370 mg·kg-1,定量限为0.001 23 mg·kg-1,可与液相色谱-质谱相媲美,同时还可避免高浓度的衍生化试剂水解产物对离子源的污染,为水中草铵膦的检测提供了一个简便、快速、准确的方法。
