Computational identifi cation and characterization of microRNAs and their targets inPenaeus monodon*
2018-07-11PULongjun濮龙军WANGJing王晶WANGYu王玉ZUOJianwei左建伟GUOHuarong郭华荣
PU Longjun (濮龙军) WANG Jing (王晶) WANG Yu (王玉) ZUO Jianwei (左建伟)GUO Huarong (郭华荣) 2
1Ministry of Education Key Laboratory of Marine Genetics and Breeding,College of Marine Life Sciences,Ocean University of China,Qingdao 266003,China
2Institute of Evolution and Marine Biodiversity,Ocean University of China,Qingdao 266003,China
AbstractStudy on shrimp miRNAs was limited and just 7 mature miRNA sequences ofMarsupenaeus japonicusare deposited in mirBase database. In this study, miRNAs and their target gene candidates were computationally identified from shrimpPenaeusmonodonand then experimentally validated. Using 39 908 expressed sequence tags (ESTs) and 21 124 genome survey sequences (GSSs) ofP.monodon(pmo) as reference dataset, a comprehensive approach based on inter-species homolog search was employed to investigate the candidate miRNAs (i.e. pmo-miRNA). A total of eight miRNAs belonging to 7 families were computationally identified and five out of them were subsequently validated by PCR and sequencing.Of these, pmo-miR-4961a, pmo-miR-4961b, pmo-miR-4979 and pmo-miR-3819 were fi rst identified from shrimps. Both the mature pmo-miRNAs and the corresponding precursors were conserved among different species. Based on perfect or near-perfect match to the target region, the target gene candidates of pmomiRNAs were predicted from 10 331 mRNA sequences ofP.monodon. A total of 20 genes were predicted as the targets of pmo-miR-4961a, pmo-miR-4961b, pmo-miR-4979 and pmo-miR-6492. Experimental validation by dual luciferase reporter assay confi rmed the targeting between 3 pmo-miRNAs and one or two of their target genes, especially the pmo-miR-4979 which could significantly down-regulate the expression of target gene (JR226772). This study updates the miRNAs and their targets inP.monodonand lays a solid foundation for future RNAi study.
Keyword:microRNA; shrimp;Penaeus monodon; target genes
1 INTRODUCTION
MicroRNAs (miRNAs) are endogenous short noncoding RNAs with 20- 23 nt in length that involve in the negative regulation of gene expression by targeting mRNAs for cleavage or translational repression based on the complementarity of the miRNA sequences to the targets (Bartel, 2004; Carthew and Sontheimer,2009). Accumulating evidences have shown that miRNAs play an important role in the cellular proliferation, differentiation and apoptosis, metabolic process and early embryo development of animals(Alvarez-Garcia and Miska, 2005; Chen, 2005). In eukaryotes, miRNA-encoding genes are widely distributed in the introns and exons of proteinencoding genes or in the intergenic regions (Saini et al., 2007). About fi fty percent of the miRNA-encoding genes have independent transcriptional promoter but the others are co-transcribed with protein-encoding genes (Rodriguez et al., 2004; Baskerville and Bartel,2005). The formation of mature miRNAs is a multistep process. First, in the nucleus, the miRNA-encoding genes are usually transcribed by RNA polymerase II as a long primary transcript called primary microRNA (pri-miRNA). Then the pri-miRNA forms a hairpin-like secondary structure,which was subsequently processed into precursor miRNA (pre-miRNA) by RNase III, Drosha. Second,the pre-miRNAs are exported by exportin-5 into the cytoplasm and further cleaved by another RNase III,Dicer into matured 20- 23 nt miRNA: miRNA*duplex, and the miRNA* is subsequently degraded.Finally, the remained single-stranded miRNA is incorporated into RNA-induced silencing complex(RISC) which mediates the subsequent posttranscriptional gene silencing (Gregory et al., 2005;Kim, 2005; Davis and Hata, 2009).
Different approaches have been adopted to identify putative miRNAs over the years. The experimental methods including the direct cloning (Kloosterman et al., 2006; Ramachandra et al., 2008), microarray (Liu et al., 2004), Northern blot and in-situ hybridization(Wienholds et al., 2005) are tedious and timeconsuming, and not successful in detecting low abundant miRNAs. Recently, rapid advances in bioinformatics and next generation sequencing (NGS)technology allowed the identifi cation of a great number of additional miRNA candidates in different organisms at low cost and high sensitivity (Bar et al., 2008; Soares et al., 2009). The computational tools developed for miRNAs identifi cation are usually based on the length,high sequence conservation among species, and structural features like hairpin and minimal folding free energy of miRNAs (Li et al., 2010; Gomes et al.,2013). The computational prediction can be used not only in species with complete genome reference sequences, but also in those without reference genomes(Li et al., 2010). For organisms whose complete genome information is not available, the expressed sequence tags (ESTs) and genome survey sequences(GSS) databases can be alternative sequence resources for the prediction of miRNA candidates and their target genes (Zhang et al., 2007; Xu et al., 2012).
With the development of various bioinformatics methods and experimental biological technology, lots of works on the identifi cation, characterization and functions of miRNAs have been done in humanHomo sapiens(Berezikov et al., 2005), model animals include mouseMusmusculus(Lagos-Quintana et al.,2002), fl yDrosophilamelanogaster(Lai et al., 2003),zebrafishDaniorerio(Kloosterman et al., 2006) and nematodeCaenorhabditiselegans(Lim et al., 2003),and model plants like thale cressArabidopsisthaliana(Adai et al., 2005). However, little attention has been paid to the miRNAs in non-model species, especially in aquatic organisms. In the past years, a small amount of miRNAs were identified and validated from big head carpHypophthalmichthysnobilisand silver carpH.molitrix(Chi et al., 2011), channel catfishIctalurus punctatus(Xu et al., 2012) and fl ounderParalichthys olivaceus(Xie et al., 2011), rainbow trout(Oncorhynchusmykiss) (Yang and He, 2014). In shrimps, seven mature miRNA and five pre-miRNA sequences identified fromMarsupenaeusjaponicushave been deposited in miRBase (Release 21, June 2014; http://www.mirbase.org/), and these miRNAs were reported to be involved in response to virus infection (Huang et al., 2012; Yang et al., 2012). 239 conserved miRNAs, 14 miRNA* sequences and 20 novel miRNAs were identified by bioinformatics analysis in white shrimp (Litopenaeusvannamei) (Xi et al., 2015). Recently, an interesting work on miRNA editing and its effect on the binding of miRNA to target gene have been reported inMarsupenaeus japonicas, which indicates the important roles of miRNA played in aquatic species (Cui et al., 2015).Penaeusmonodonis an economically important aquaculture species with the largest farming area and breeding production in cultured shrimps (Rengpipat et al., 1998). Information on miRNAs and their target genes inP.monodonis also very limited, however, a computational workf l ow has been developed to identify conserved miRNA genes in the 10 536 uniqueP.monodonESTs (Meemak et al., 2013).
The main purpose of the present study was to identify miRNAs candidates and their potential target genes from the known EST and GSS databases ofP.monodonusing comparative algorithms. The results obtained could guide the future experimental validations and contribute to the understanding of the roles of these miRNAs in regulating the development and metabolism ofP.monodon.
2 MATERIAL AND METHOD
2.1 Cells and culture
The mammalian Neuro-2a cell, purchased from ATCC cell bank, was a fast-growing mouse neuroblastoma cell line. Neuro-2a cells were maintained in Dulbecco's modified Eagle’s medium(DMEM, Life Technologies, USA) supplemented with 10% bovine calf serum (BCS, Life Technologies,USA), and cultured at 37°C in a 5% CO2incubator.
2.2 miRNA reference set

Fig.1 Flowchart of miRNAs prediction procedure in P. monodon
In order to increase the accuracy of prediction,only the published miRNAs from thephylumarthropoda, close to shrimps in relationship, were selected as references for the identifi cation of potential miRNA homologies inP.monodon. A total of 2 669 previously identified mature miRNAs and their premiRNAs sequences were obtained from miRNA registry Database (Release 20, June 2013; http://www.mirbase.org/). These miRNAs were defi ned as a reference set of miRNA sequences. To avoid the redundant or overlapping miRNAs, the repeated sequences of miRNAs within the above reference set were removed by Perl scripts (http://www.perl.org)and the remaining sequences were used as query sequences for BLAST search. These known miRNAs references are from 17 species including shrimpM.japonicas, water fl eaDaphniapluex, mosquitoAedesaegypti, butterf l yHeliconiusmelpomene, mothManducasexta, miteTetranychusurticae, aphidAcyrthosiphonpisum, acrididLocustamigratoria,beetleTriboliumcastaneum, silkwormBombyxmori,two ticks ofIxodesscapularisandRhipicephalus microplus, two bees ofApismelliferaandNasonia vitripennis, and three fl ies ofDrosophila melanogaster,D.pseudoobscuraandD.simulans,
2.3 Data sources of expressed sequence tags (EST),genome survey sequences (GSS) and mRNAs of P.monodon
EST, GSS, and mRNA sequences ofP.monodonwere downloaded from GenBank nucleotide database of National Center for Biotechnology Information(NCBI, http://www.ncbi.nlm.nih.gov/). A total of 39 908 EST sequences and 21 124 GSS sequences have been obtained and used as the searching sources for miRNA. A total of 10 331 mRNA sequences were obtained for the searching sources for target genes. To avoid redundancy, the downloaded EST, GSS and mRNA sequences were blasted against each other and the sequences with high similarity (>98%) were removed.
2.4 Prediction of potential miRNAs
The unique miRNA reference set was used as query sequences to blast against theP.monodonEST and GSS databases. The BLAST search was carried out through BLAST 2.2.28+ (Zhang et al., 2000). The sequences which had less than 3 nt mismatch with query miRNA sequences and more than 16 nucleotides in length were manually chosen as potentialP.monodonmiRNAs. The blast parameter settings were adjusted as follows: an expect value cut-off of 10, a low-complexity sequence fi lter, 1 000 descriptions and alignments, and automatically adjusted parameters for short input sequences to improve the accuracy of blast result. Figure 1 showed the flowchart of miRNAs prediction procedure inP.monodon.
2.5 Secondary structure analysis of predicted P.monodon miRNAs (pmo-miRNAs)
All the predicted mature miRNA sequences along with their 200 bp upstream and 200 bp downstream fl anking sequences were selected from theP.monodonETS and GSS databases and subjected to RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi ) to predict and generate the secondary structure with the default parameters (Hofacker,2003). In each case, we chose the lowest-energy secondary structure as the target architecture for the next validation. Only the sequences which fi t the following strict criteria were regarded as the potential miRNAs and pre-miRNAs inP.monodon: (1) the potential miRNA sequence did not contain more than 3 nt mismatches with the query homology; (2) the selected pre-miRNA could be folded into stem-loop hairpin secondary structure and the mature miRNA sequence was not located in the terminal loop of the hairpin structure; (3) the mismatches between miRNA and the complementary sequence (miRNA*) was less than 6 nt; (4) the potential mature miRNAs should be located in the same arm of the stem-loop hairpin structure with their known homologs; (5) the potential pre-miRNA had higher minimal folding free energy index (MFEI) than other non-miRNAs. MFEI was calculated by the following formula: MFEI=[(minimal folding free energy/length of RNA sequence)×100]/(G+C)%; (6) the potential miRNA sequence could not contain large loops or breaks in microRNA:microRNA* duplex (Yin et al., 2008; Wang et al.,2012). Finally, all the selected pre-miRNA could be folded into stem-loop hairpin secondary structure and have higher minimal folding free energy index(MFEI) than other non-miRNAs. Thus the length of the actual miRNAs precursor might be longer than what is presented here.
2.6 Phylogenetic analysis of the identified pmomiRNAs
Both the pre-miRNAs and mature miRNAs sequences are highly conserved among different animal species, thus homology analysis of the identified pmo-miRNAs can reveal their evolutionary relationships. The identified mature pmo-miRNAs and pre-pmo-miRNAs were aligned with the known miRNAs and pre-miRNAs from the same miRNA family but different species, respectively, and the corresponding evolutionary trees were generated by maximum likelihood (ML) method (Strimmer and von Haeseler, 1996) following 1 000 bootstrapped replicates and other default parameters. All the analyses were performed on the MEGA 5.2 software(Tamura et al., 2011).
2.7 Prediction of the pmo-miRNA targets
The widely used computational algorithm of miRanda was employed in this study to predict the potential target genes of pmo-miRNA (John et al.,2004; Griffths-Jones et al., 2006). To minimize false positives, two sets of criteria were used for the screening of pmo-miRNA targets and only the target sites predicted by both sets of criteria were accepted as the potential target genes. One criteria were set as followed: (1)S, the sum of single-residue-pair match scores over the alignment, ≥ 140; (2) ΔG, the free energy of duplex formation, <-17 kcal/mol. The other criteria were set as followed: (1) the number of mismatches between the predicted miRNA and complementary target sequence was not beyond four(G-U base pair was counted as 0.5 mismatches); (2)two consecutive mismatches did not exist in the miRNA: target duplex; (3) mismatches occurred in the fi rst 9 bp of the 5'end of miRNA in the miRNA:target duplex were not more than one; (4) positions 10 and 11 (numbered from the 5'end of miRNA) in the miRNA : target duplex, which were considered as the cleavage sites, should not contain any mismatch; and(5) no gap appeared in the miRNA: target duplex(Wang et al., 2012). Target sequences fi ltered by the above criteria were further fi ltered by BLASTX search program to remove the targets without significant similarity and then the remaining target sequences were subjected to functional assignment.
2.8 PCR validation of the pmo-miRNAs
To validate the computationally identified pmomiRNAs, polymerase chain reaction (PCR) was performed to isolate each of the predicted pmomiRNAs from the cDNA templates ofP.monodon.The primers for amplifying miRNAs were designed according to the primary sequence and located in the each fl ank of mature miRNAs. Primers used for amplifying the 8 pmo-miRNAs are listed in Table 1.
Total RNA was extracted from the shrimp muscle using Trizol reagent (TransGen, Beijing, China) and then digested with RNase-free Dnase I (TaKaRa,Otsu, Japan) in the presence of RNase inhibitor(TaKaRa, Otsu, Japan) according to the manufacturer’s protocol. First-stranded cDNA was synthesized in20 μL reaction volume by ReverTra Ace qPCR RT Kit(Toyobo, Japan) with 2 μg of total RNA as template and 1 μL oligo (dT) as primer. PCR was performed in 20 μL total volume consisting of 2.0 μL 10× PCR buffer (with Mg2+), 1.6 μL 2.5 mmol/L dNTPs,1.0 μL 10 μmol/L forward primer, 1.0 μL 10 μmol/L reverse primer, 0.2 μL 5 IU/μL Taq DNA polymerase, 1 μL cDNA template and 13.2 μL ddH2O. One cycle of 94°C for 10 min, 32 cycles of 94°C for 45 s, 50-60°C annealing for 45 s and 72°C for 50 s, followed by one cycle of 72°C for 10 min. The PCR results were analyzed by 1.5% agarose gel electrophoresis.

Table 1 Primers used for amplifying the 8 pmo-miRNAs
2.9 GO and KEGG analysis
To further understand the functions and metabolic pathway of target genes, gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)analyses were performed using Blast2go suit(Ashburner et al., 2000; Kanehisa and Goto, 2000;Conesa et al., 2005; Götz et al., 2008). First, all the target genes were searched against non-redundant(nr) database and annotated by BLASTX with the e-value of 1e-6 (Altschul et al., 1997). The functional category for each target gene was annotated according to the best hits results generated in the mapping of Gene Ontology (GO) terms with default parameters.The metabolic pathway participated by each target gene was analyzed by searching KEGG database.
2.10 Experimental validation of the targeting activity of 3 pmo-miRNAs to their target genes
Dual luciferase reporter gene assay system (Galen Biopharm International Co. Ltd., Beijing, China) was used to experimentally validate the targeting activity of 3 PCR-confi rmed pmo-miRNAs to their predicted target genes. Based on the computational prediction results mentioned above, three pmo-miRNAs and their corresponding two target genes each were randomly selected as followed: miR-4961a and its target genes of JR227957 and JR227699, miR-4961b and its target genes of JR227701 and JR226950, miR-4979 and its target genes of JR226772 and JR225956.Three methylated single-strand miRNA mimics of miR-4961a (5'-UCUUUGUGUGUAUGUAUGUAU-3'), miR-4961b (5'-UCUGUAUGUCUAUGUAUGUAU-3') and miR-4979 (5'-UACAUGUGUGUGUAUAUAUAU-3') and one negative control mimic(5'-UCACAACCUCCUAGAAAGAGUAGA-3') were synthesized by Shanghai GenePharma Co. Ltd., China.
All sequences of the 6 selected target genes were extracted from EST-GSS database and the corresponding sequences of 52 bp fragment in length fl anking the predicted targeting site was truncated,respectively. And then for each of the 6 truncated fragments, a pair of complementary single-strand oligonucleotides with an addition of XhoI or NotI sites at both ends, 59 bp in length each, were synthesized by BGI company (Shenzhen, China),respectively, and then subjected to 5'-end addition of phosphate and annealing reaction to form doublestrand insertion unit. The annealing reaction was performed in 20 μL total volume consisting of 8 μL 0.1 nmol/μL oligonucleotide for each strand, 0.5 μL 10 mg/mL ATP-Na2, 0.4 μL 10 U/μL T4 polynucleotide kinase (PNK), 2 μL 10×T4 PNK buffer and 1.1 μL ddH2O and then incubated at 37°C for 30 min,followed by 95°C for 5 min and cooled to room temperature. After that, the obtained double-strand oligonucleotide was directionally inserted into a reporter plasmid of psiCHECK-2 (kindly provided by Jerame Hui, School of life science, Chinese University of Hong Kong, China). The ligation reaction mix in 15 μL total volume included 10 μL of the obtained double-strand oligonucleotide, 0.7 μL 0.5 μg/μL linear psiCheck-2 plasmid DNA, 1 μL 350 U/μL T4 ligase, 1.5 μL 10×T4 ligase buffer and 1.8 μL ddH2O.At the same time, for each of the 6 target genes tested,a mutant double-strand oligonucleotide with mutations introduced in the regions corresponding to the seed sequences of pmo-miRNAs was also preparedin the same way as mentioned above, respectively. All the synthesized wild-type and mutant target sequences were listed in Table 2.

Table 2 Oligonucleotide sequences used to form double-strand targeting fragments
One day prior to transfection, Neuro-2a cells were seeded into each well of a 24-well culture plate.Approximately 60%-70% conf l uence was anticipated at the time of transfection. All the stock solutions of 12 psiCHECK-2 recombinant plasmids containing wild-type (WT) or mutant target gene regions, 3 synthesized single-strand miRNA mimics and 1 synthesized negative control mimic were prepared in RNase-free water to a concentration of 25 μmol/L.Then to validate the targeting between pmo-miRNAs and their target genes tested, varied psiCHECK-2 recombinant plasmid DNAs with the same concentration of 250 ng/μL wild-type or mutant target gene regions were co-transformed with their corresponding miRNA mimic or negative control mimic (25 μmol/L) into the Neuro-2a cells using Lipofactamine 2000 transfection reagent (Invitrogen)following the product manual. The following groups were tested: 1: negative control mimic (1.6 μL) + psi-CHECK2-WT (4 μL); 2: miRNA mimic (1.6 μL) +psiCHECK2-WT (4 μL); 3: negative control mimic(1.6 μL) + psiCHECK2-mutant (4 μL); 4: miRNA mimic (1.6 μL) + psiCHECK2-mutant (4 μL). For each group, all volumes were multiplied by 3.5 to account for the triplicate samples and liquid loss during pipetting.
At 48 h post-transfection, the old medium in each well was removed and the cells were rinsed with PBS twice. Then 150 μL per well of cell lysis buffer was added and the culture plate was shaken gently at room temperature for 15 min to ensure complete lysis. After that, all the cell lysate in each well was transferred into a 1.5-mL centrifuge tube and centrifuged at 9 000×gfor 5 min. Then 20 μL of cell lysate supernatant was taken out and added into another 1.5 mL centrifuge tube containing 100 μL of luciferase assay reagent and mixed quickly by pipetting two or three times. Then the tube was immediately placed in the luminometer (GloMax, Promega) and the luminescence (relative light units, RLU) due to fi refly luciferase activity was measured. After that, the sample tube was removed from the luminometer and 100 μL of stop reagent was added and mixed brief l y,then the sample tube was put in the luminometer again, and the luminescence due to Renilla luciferase activity was recorded. As the Renilla luciferase gene was set as reporter gene and fi refly luciferase gene was set as inner reference in the plasmid map of psiCHECK2, the ratios of Renilla luciferase activity to fi refly luciferase activity was calculated for each sample and used to detect the targeting activity between pmo-miRNAs and target genes. The SPSS software was used for data statistical analysis.
3 RESULT AND DISCUSSION
3.1 Eight miRNA candidates were identified from P. monodon
A total of 2 669 known miRNAs were collected from 17 different species of phylum arthropoda,which were in close relationship withP.monodon.Multiple sequence alignment removed 485 repeat miRNAs from this data set. The remaining 2 184 nonredundant miRNAs were blasted individually as query sequence against ESTs (39 908 sequences) and GSS (21 124 sequences) ofP.monodonusing local BLAST-2.2.28+. The results obtained showed that,155 sequences (70 from EST and 85 from GSS) had less than 3 nt mismatches with the known query miRNAs and selected for subsequent miRNAs identifi cation. From these selected sequences, 37 protein-encoding sequences were further removed by BLASTX program. After that, the secondary structures of the remaining 118 sequences were predicted and generated by RNAfold web server, and then they were screened for potential miRNAs by evaluating the secondary structures and sequences based on the criteria described in Methods. Finally, a total of 8 potential miRNA sequences (4 from EST and 4 from GSS) were obtained fromP.monodon.
The sequences and hairpin structures of the identified eight miRNAs were shown in Fig.2. They belong to 7 different miRNA families, named as pmomiRNA4961a, pmo-miRNA4961b, pmo-miRNA4979,pmo-miRNA6489, pmo-miRNA6491, pmo-miRNA6492,pmo-miRNA6493 and pmo-miRNA3819. These miRNA families have been reported previously in other animals, indicating the functional conservation of miRNA members in these families.
3.2 Characterization of the predicted P. monodon miRNAs (pmo-miRNAs)
The identified pmo-miRNAs were diverse not only in the variety of miRNA families they belong to, but also in the length of pre-miRNAs, (A+U) contents and location of mature miRNA sequence within their pre-miRNAs. For all the identified pmo-miRNAs, the length of pre-miRNAs varied from 59 nt to 143 nt, but the precursors containing 60-95 nt accounted for 87.50%. In contrast, the lengths of the majority of the plant pre-miRNAs were more consistent, typically 70-80 nt (Yin et al., 2008). The (A+U) contents in each pre-pmo-miRNAs ranged from 26.60% to 83.33%, which conforms to the standard that miRNA precursors should have 30%-70% (A+U) contents.MFEI (minimal folding free energy index) is an important parameter to distinguish miRNAs from other types of RNAs. As shown in Table 3, the MEFI values for all the identified pmo-miRNAs ranged from 0.60 to 4.07, which was in agreement with previous studies (Zhang et al., 2006a). Of the identified eight pmo-miRNAs, six pmo-miRNAs have mature sequences located in the 5'end of their secondary hairpin structures, and the other two in the 3'end instead. And also, three mature sequences started with 5'uracil, indicating an important role of uracil in the regulatory function of miRNAs mediated by RNA-induced silencing complex (RISC) (Bartel,2004; Gregory et al., 2005; Kim, 2005).
As shown in Table 3, about 0-3 nt base substitutions exist in the mature miRNA sequences betweenP.monodonand other known miRNAs, showing both conservation and divergence of miRNAs in evolution(Zhang et al., 2005). Although animals have more non-conserved miRNAs than plants (Axtell et al.,2011), animal miRNAs are still highly conserved among different species and a large amount of miRNA candidates can be found using homological search methods. But Zhang et al. (2006b) reported a low prediction frequency of miRNAs from EST using comprehensive bioinformatics approach as 0.01%.Similar result was obtained in this study. Only 8 miRNA candidates were predicted from 61 132 EST sequences, the miRNA prediction frequency was about 0.013%. Two reasons may account for the above low prediction frequency: (1) miRNAs were transcribed from either sense or antisense strand ofDNA at the miRNA genomic loci (Stark et al., 2008).miRNAs located in the complementary strands of EST sequences have been missed; (2) in order to improve the reliability of prediction, only known miRNAs from 17 species of the phylum arthropoda,close to shrimps in relationship, were used as reference query for blast in this study. This markedly decreased the miRNA prediction frequency, too. Despite all that,the reliability of the predicted pmo-miRNAs could be high because they are screened by combination of stringent criteria and secondary structure prediction.

Table 3 Characterization of the identified pmo-miRNAs

Fig.2 Sequences and harpin sturctures of the identified miRNAs from P. monodonThe mature miRNA sequences are underlined and in red colour. The length of the actual miRNAs precursor might be longer than what is presented here.
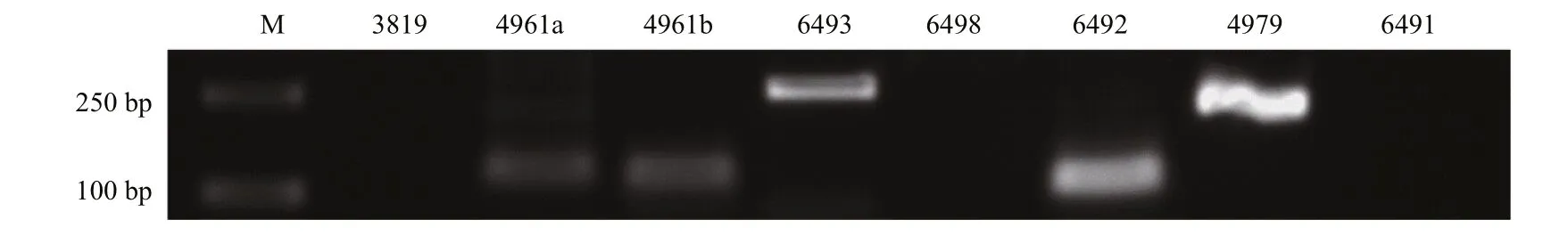
Fig.3 PCR validation of the identified 8 pmo-miRNAsM: molecular weight marker 2 000.
Works on the whole genome sequencing of shrimp are going on. When more genomic information of shrimp is publicly available, more shrimp miRNAs will be discovered using both classic experiment methods and bioinformatics approaches.
3.3 PCR validation of the predicted pmo-miRNAs
PCR was performed to experimentally validate the 8 pmo-miRNAs predicted using Specific primers fl anking the pre-pmo-miRNAs sequences and the obtained PCR products were separated on a 1.5%agarose gel. As shown in Fig.3, five computationally predicted pmo-miRNAs including pmo-miR-4961a,pmo-miR-4961b, pmo-miR-4979, pmo-miR-6492 and pmo-miR-6493 were successfully amplified from the cDNA templates of muscle tissues ofP.monodonwith the expected product sizes (Table 1). All the amplified fragments were also successfully verified by sequencing, indicating a 100% positive confi rmation. However, other three pmo-microRNAs precursors were not detected using PCR. Limited tissues sampled and inappropriate PCR reaction condition possibly accounted for the failure.
3.4 Phylogenetic analysis of the identified pmomiRNAs
To show the evolutionary relationship of both intra- and inter-family of miRNAs, all the mature pmo-miRNAs or pre-pmo-miRNAs predicted were included in the construction of phylogenetic trees(Fig.4). As shown in the phylogenetic tree for mature pmo-miRNAs (Fig.4a), five mature pmo-miRNAs were found to have highly conserved counterpart miRNAs from different species and clustered in the same position, e.g. pmo-miR-3819 and tca-miR-3819-5p, pmo-miR-6489 and mja-miR-6489-3p,pmo-miR-6493 and mja-miR-6493-5p, pmomiR-6491 and mja-miR-6491, pmo-miR-6492 and mja-miR-6492. However, pmo-miR-4979 and dmemiR-4979-3p were clustered in the same branches but different evolutionary distances. Ofinterest, pmomiR-4961a and pmo-miR-4961b were from the same species and same precursor but deposited in different branches, showing a divergent evolution rate in the tree. It can be concluded that different miRNAs might evolve at different rates in both inter- and intraspecies.
Rodriguez et al. (2004) suggested that pre-miRNA sequences were conserved, too. Similar results were obtained in this study. Unlike the mature pmomiRNAs, in the phylogenetic tree for the pre-pmomiRNAs, pre-pmo-miR-6489 and pre-mja-miR-6489 were clustered in the same branch but with different evolutionary distances, pre-pmo-miR-3819 and pretca-miR-3819 were located in completely divergent branch, indicating that the fl anking sequences outside the core mature sequence were relatively less conserved than the mature sequence in the miRNA.
The phylogenetic trees for mature and precursor pmo-miRNAs also revealed that four of the identified pmo-miRNAs have already been discovered inM.japonicus(Huang et al., 2012), a closely related species toP.monodon, e.g. mja-miR-6489, mjamiR-6491 mja-miR-6492 and mja-miR-6493. This confi rmed to some degree the reliability and accuracy of computational identifi cation of miRNAs. Of course, there are some miRNA members found inM.japonicusbut not predicted in this study, referring that they might not be deposited in the EST or GSS datasets yet. Thus, deep sequencing and genome-wide identifi cation are needed to help to solve this problem in the future (Bar et al., 2008; Lu et al., 2008; Zhou et al., 2010).
3.5 Prediction of the target genes of the 8 pmomiRNAs
Identifi cation of the target genes of the 8 pmomiRNAs is an easy way to gain an insight into their roles in various cellular functions and gene regulation network. It has been reported that miRNAs always perfectly or near-perfectly match to their target mRNAs and regulate gene expression in posttranscriptional level by inhibiting the protein translation or facilitating mRNA degradation (Brown and Sanseau, 2005; Sethupathy et al., 2006), and during this process only the evolutionarily conserved mature miRNA sequences seem to be necessary and thus often used in the target gene identifi cation. Based on the complementary between miRNAs and targets,putative target genes for the 8 pmo-miRNAs were identified using miRanda software under stringent criteria. As shown in Table 4 and Fig.5, only 4 pmomiRNAs of pmo-miR-4961a, pmo-miR-4961b, pmomiR-4979 and pmo-miR-6492 successfully matched to 20 target genes after searching against the downloaded mRNA database ofP.monodonfrom NCBI. This suggested that one pmo-miRNA could target multiple genes inP.monodon, too. These different genes targeted by one pmo-miRNA often had quite different biological functions and were even involved in different cellular process and metabolic pathways. The mechanism of action for this is still unclear and more works are needed. The remaining 4 pmo-miRNAs of pmo-miR-3819, pmo-miR-6489,pmo-miR-6491 and pmo-miR-6493 failed to match annotated target genes instead. The incompleteness of mRNA database ofP.monodonmay account for the failure.
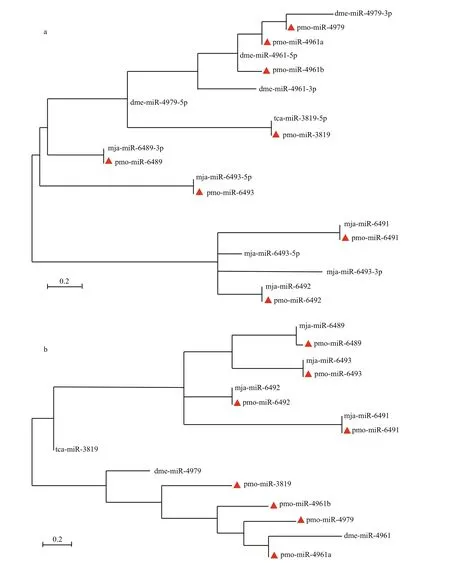
Fig.4 Phylogenetic analysis of mature pmo-miRNAs (a) and pre- pmo-miRNAs (b)identified pmo-miRNAs are labeled with red triangle. Other published miRNAs from the same families of the 8 pmo-miRNAs are from species ofTribolium castaneum(tca),Drosophila melanogaster(dme) andMarsupenaeus japonicus(mja), respectively. Precursors of dme-miR-4961, dme-miR-4979, tcamiR-3819, mja-miR-6489 and mja-miR 6493 can produce two mature miRNAs. They are dme-miR-4961-5p and dme-miR-4961-3p, dme-miR-4979-5p and dme-miR-4979-3p, tca-miR-3819-5p and tca-miR-3819-3p, mja-miR-6489-5p and mja-miR-6489-3p, mja-miR-6493-5p and mja-miR-6493-3p, respectively.

Table 4 Potential target genes ofidentified pmo-miRNAs
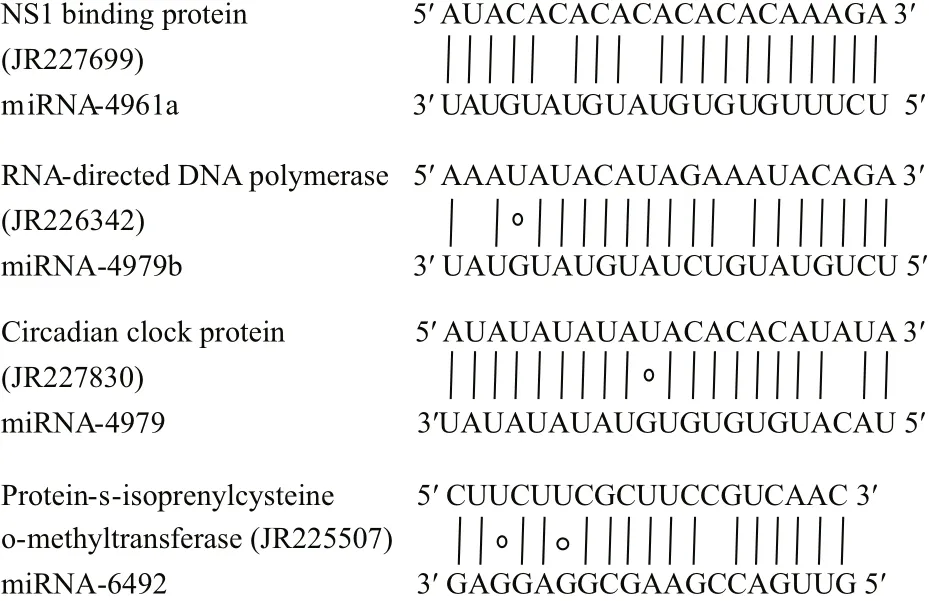
Fig.5 Representative matches between pmo-miRNAs and their target genesTop strands show the pmo-miRNA binding sites in the target genes,and the bottom strands shows the pmo-miRNAs. Watson-Crick pairing(vertical dashes) and G:U wobble pairing (circles) are indicated.
Many genes associated with transcription process were reported as the targets of miRNAs in both animals and plants (Guo et al., 2009). In this study, we found that pmo-miR-4961, a targeted RNA polymerase I-Specific transcription initiation factor ofP.monodon(JR227957), indicating its role in the mRNA transcription.
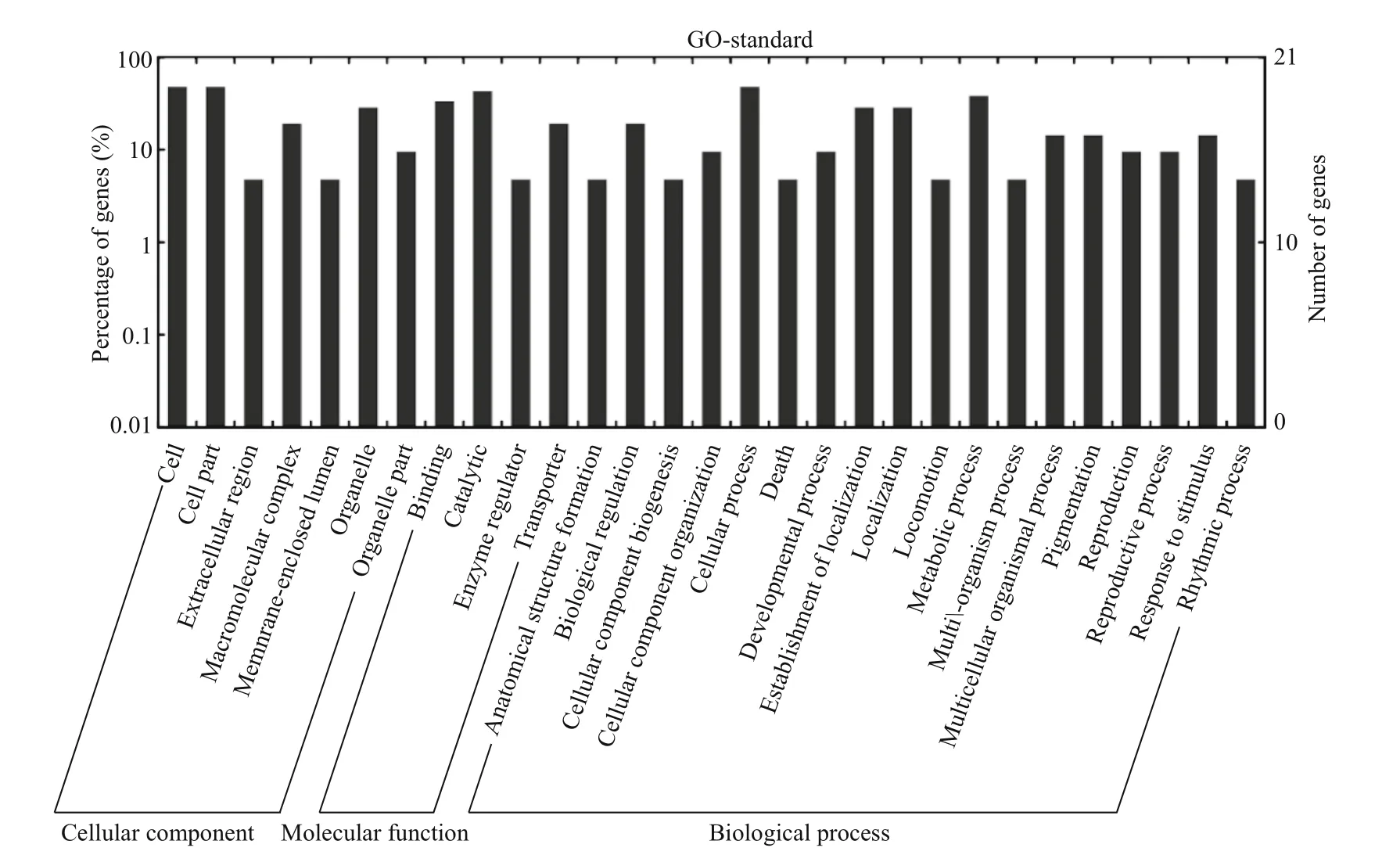
Fig.6 Gene ontology categories and distribution of pmo-miRNAs target genes
The growth and development of animals are an intrinsic process in which many signal pathways and gene regulation networks were involved. Previous studies have demonstrated that miRNAs played an important role in the regulation of growth,development and apoptosis (Alvarez-Garcia and Miska, 2005; Cheng et al., 2005). Here, the pmomiR-4979 was found by both computational prediction and experimental validation to target the gene of crustacean hyperglycemic hormone (CHH)precursor (JR226772, Table 4 and Fig.7). CHH protein, a member of neuropeptide hormone family present only in arthropods, plays a pivotal role in the modulation of hemolymph glucose levels, molting,reproduction and the stress response. It regulates the growth, reproduction and molting of crustacean by interacting with molt-inhibiting hormone (MIH),gonad-inhibiting hormone (GIH) and mandibular organ-inhibiting hormone (Chan et al., 2003; Fanjul-Moles, 2006). The target of pmo-miR-4979 to CHH gene inferred an involvement of this pmo-miRNA in the growth and molting ofP.monodon. Circadian clock protein gene, which responds to light stimulus,was also identified as target of pmo-miR-4979,suggesting another function of this miRNA in the regulation of cellular response to light/dark. Up to date, several miRNAs have been previously reported in the sensory cells and organs (Ason et al., 2006; Li et al., 2006; Weston et al., 2006), for example,miR183 has been reported to be expressed conservatively in epithelial cells ofS.kowalevskiiand sensory organs of D. melanogaster and nematode(Pierce et al., 2008).
Ofinterest, cAMP-dependent protein kinase type 1 regulator subunit (JR226950) and protein phosphatase(JR228373) were also identified as target genes for pmo-miR-4961b and pmo-miR-4979, respectively.The above two enzymes act as opposite regulators for the activity of target proteins by phosphorylation and dephosphorylation (Fabbri et al., 2007; Kato et al.,2009; O’Connell et al., 2009). This means that pmomiR-4961b and pmo-miR-4979 may participate in the functional regulation of the same protein target in the signaling pathways via knocking down the activity of kinase and phosphatase, respectively. And also, RNA-directed DNA polymerase (JR226342) was found as the target of pmo-miR-4961b. As we know, the infection and replication of RNA virus in host cells greatly depend on this enzyme. Shrimp RNA virus like Taura syndrome virus (TSV) is a big threat to shrimp industry, thus pmo-miR-4961b might provide us a useful tool to treat TSV infection.
3.6 GO classifi cation and KEGG pathway analysis of the predicted 20 target genes
To further understand the cellular functions and regulatory pathways involved by pmo-miRNAs, all the predicted target genes were subjected to GO and KEGG database for analysis. GO terms were categorized into three classes of cellular component,molecular function and biological process (Ashburner et al., 2000). As shown in Table 5, GO analysis showed that the 20 pmo-miRNA target genes were assigned to 7 categories at cellular component level,4 categories at molecular functions level and 18 categories at biological process level. Although number of pmo-miRNA targets predicted in this study is limited, the distribution of assigned GO terms was also produced by Wego output to show gene function visually and deeply (Fig.7). As shown in Fig.6, more pmo-miRNA targets were located in cell and cell part, followed by those located in organelle and macromolecular complex. All putative target genes presented a narrow range of molecular function including binding, catalytic, enzymeregulator and transporter. The biological process involved by the pmo-miRNA targets includes cellular process, establishment of location and metabolic process, etc. Several target genes of pmo-miRNAs were identified in the process of development,reproduction and death. This enriched the known miRNA library (Carrington and Ambros, 2003;Kloosterman and Plasterk, 2006). The results of KEGG annotation have shown that pmo-miRNA targets participate in many pathways, such as terpenoid backbone biosynthesis, apoptosis, insulin signaling pathway and thyroid hormone signal pathway (Table 6). Information on the metabolic networks of pmo-miRNA targets will help us to understand the functions and target pathways of pmo-miRNAs in more detail and guide the future research ofinterest.
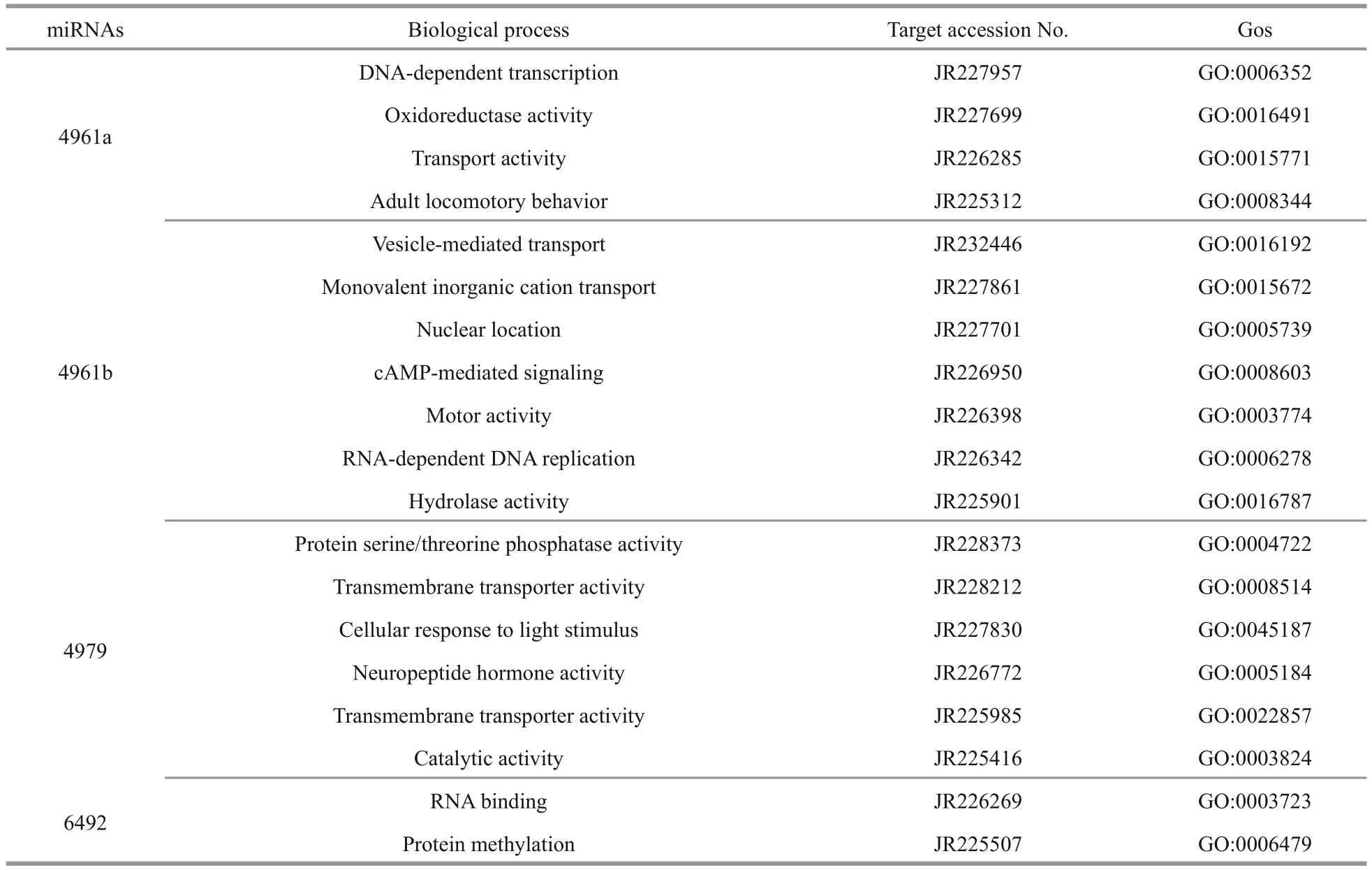
Table 5 Biological process analysis of the pmo-miRNA target genes

Table 6 Pathway annotation for pmo-miRNAs targets in KEGG
3.7 Experimental validation of the targeting activity of 3 pmo-miRNAs to their target genes
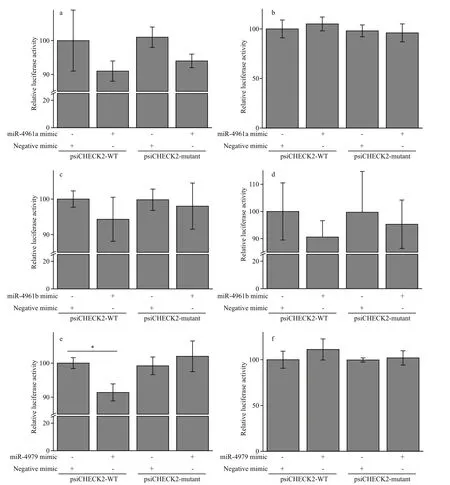
Fig.7 Experimental validation of the targeting of 3 pmo-miRNAs to their corresponding target genes in Neuro-2a cells by dual luciferase reporter assaya. miRNA-4961a and its target gene (JR227957); b. miRNA-4961a and its target gene (JR227699); c. miRNA-4961b and its target gene (JR227701);d. miRNA-4961b and its target gene (JR226950); e. miR-4979 and its target gene (JR226772); f. miR-4979 and its target gene (JR225956); All data are expressed as mean±SE (n=3). One-way ANOVA in SPSS was used for statistical analysis. F02A indicates significant difference (P<0.05).
The targeting activity of 3 PCR-confi rmed pmomiRNAs of pmo-miR-4961a, pmo-miR-4961b and pmo-miR-4979 to their predicted gene candidates were investigated in Neuro-2a cells by dual luciferase reporter gene assay. As shown in Fig.7, obvious down-regulation of the expression of target genes was found in experimental groups of pmo-miR-4961a and JR227957, pmo-miR-4961b and JR227701, pmomiR-4961b and JR226950, pmo-miR-4979 and JR226772. However, significant targeting activity was found only between pmo-miR-4979 and JR226772 (P<0.05). The target gene JR226772 encoded crustacean hyperglycemic hormone precursor, suggesting the involvement of pmomiR-4979 in the regulation of neuropeptide hormone activity. However, the remaining three experimental groups failed to detect obvious targeting activity,indicating again that prediction data based on computational algorithm need to be experimentally validated to avoid potential false-positive result. In this study, a mammalian cell line was used in the validation experiment of miRNA-mRNA interaction due to the absence ofimmortalized shrimp cell line.Difference in genetic background between these two cell lines might to some degree account for the failure mentioned above. The mechanism of miRNA-mediated gene silencing is very different between vertebrate and invertebrate. In vertebrate, a miRNA is loaded onto Argonaut 2, while a miRNA is loaded onto Argonaut 1 in invertebrate.
4 CONCLUSION
In this study, a total of 8 miRNAs were computationally identified from EST or GSS database ofP.monodon. Of these, pmo-miR-6489, pmomiR-6491, pmo-miR-6492 and pmo-miR-6493 have been reported previously inM.japonicus, but pmomiR-4961a, pmo-miR-4961b, pmo-miR-4979 and pmo-miR-3819 were fi rst identified in shrimps. And also five out of the eight predicted pmo-miRNAs including pmo-miR-4961a, pmo-miR-4961b, pmomiR-4979, pmo-miR-6492 and pmo-miR-6493 were subsequently validated by PCR and sequencing.Experimental validation by dual luciferase reporter assay confi rmed the targeting between 3 pmomiRNAs and one or two of their target genes,especially the pmo-miR-4979 which could significantly down-regulate the expression of target gene (JR226772). This study updates the miRNAs library and their targets inP.monodonand lays a solid foundation for future RNAi study.
5 ACKNOWLEDGEMENT
We thank Dr. Jerame Hui (School of Life Science,Chinese University of Hong Kong, China) for kindly providing the reporter plasmid of psiCHECK-2.
猜你喜欢
杂志排行

Journal of Oceanology and Limnology的其它文章
- Response of the North Pacific Oscillation to global warming in the models of the Intergovernmental Panel on Climate Change Fourth Assessment Report*
- Effect of mesoscale wind stress-SST coupling on the Kuroshio extension jet*
- Surface diurnal warming in the East China Sea derived from satellite remote sensing*
- Cross-shelf transport induced by coastal trapped waves along the coast of East China Sea*
- Observations of near-inertial waves induced by parametric subharmonic instability*
- Seasonal variation and modal content ofinternal tides in the northern South China Sea*