基于大量ChIP数据集的果蝇顺式调控模块的从头预测
2018-05-07张少强
李 婷,张少强
(天津师范大学 计算机与信息工程学院,天津 300387)
随着新技术的快速发展,基因组测序的成本下降,特别是转录因子的ChIP-seq技术的广泛使用[1],使得很多后生动物和植物产生了海量的ChIP-seq数据集.尽管目前已有大量的预测顺式调控元件和模块的工具,但在大型基因组中,整合指数级增长的ChIP数据集,并在全基因组范围预测顺式调控元件和模块,却一直是具有挑战性的计算问题[1-4].一定数量的转录因子常常组合起来,共同调控不同细胞类型、组织、发育阶段和生理条件下的不同基因[5],与这些共同调控的转录因子相结合的非编码DNA位点(即顺式调控元件)构成了其顺式调控模块.大量的ChIP数据集中包含着一定的模块组合信息,这些信息是由不同转录因子共同转录调控而形成的[6-7].因此,利用不同细胞类型、组织、发育阶段和生理条件下的不同转录因子的大量ChIP数据集,就有可能通过对模体进行整合以寻找共现模式,进而对某种真核生物全基因组范围顺式调控模块进行从头预测.
本文基于果蝇已有的ChIP数据集,采用模体发现算法FisherNet及高性能并行的模体聚类算法CLIMP对果蝇的顺式调控模块进行从头预测,并与较新的DePCRM算法[8]进行了比较.本文研究方法的流程如图1所示.

图1 顺式调控模块预测流程图Fig.1 Flow chart of predicting CRMs
1 数据来源与预处理
1.1 数据来源
由于果蝇常被用来研究动物基因的转录调控,大量的顺式调控元件和模块已被实验验证,而且在过去的几年中该生物已经产生了大量的ChIP-chip和ChIP-seq数据,因此本文使用果蝇作为模式生物评估算法.为此,整理了来自56个不同转录因子的168个ChIP-chip和ChIP-seq数据集,这些数据集包含不同的发育阶段(胚胎、幼虫期1~3、蛹和雌雄成虫)和不同实验条件下(热休克等)的结果,其中:42个ChIP-chip和42 个 ChIP-seq 数据集来自 modENCODE 项目[6,9],38个ChIP-chip数据集来自Berkeley果蝇转录网络项目(BDTNP)[10],46个ChIP-chip数据集来自文献[8].
1.2 数据预处理
利用peak-calling工具[11]查找ChIP数据中结合峰的序列,这些序列包含丰富的对应转录因子的顺式调控元件.将较短的结合峰从两端延伸到3 000个碱基长的序列(这个长度与典型顺式调控模块的长度相当),使得结合最高峰正好位于序列中部.除了ChIP实验的转录因子的顺式调控元件外,扩展的结合峰更可能包含辅助调控转录因子(在顺式调控模块中共同作用的转录因子)的顺式调控元件.
2 算法步骤
数据预处理后的具体算法流程见图2.
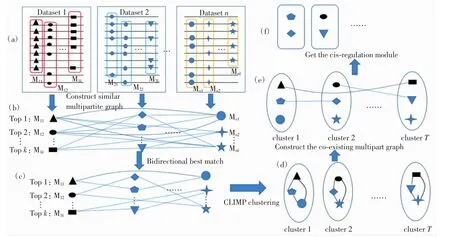
图2 数据预处理后的具体算法流程图Fig.2 Flow chart of detailed algorithm after data preprocessing
2.1 构建模体相似多部图
对于每组延伸后的结合峰序列数据集,运用模体发现工具FisherNet算法[12]寻找大量的假定模体.对每个数据集输出前k个最优的模体,见图2(a),k默认值为20.
对于预处理的每个数据集输出的前20个最优模体,以每个模体做为顶点,考虑到2个模体的频率矩阵和位置权重矩阵,本文使用位置信息含量相似度量法 SPIC(similarity with position information contents)[13]计算不同数据集间模体的相似性(阈值为0.7),SPIC度量法已被证实优于其他度量公式[13],若2个模体的相似度大于阈值,则连接2个模体,从而构建模体相似多部图,见图2(b).数据集内部模体之间不连边,只计算不同数据集间模体的两两相似性.
构建模体相似多部图后,运用双向最佳匹配BDBM(bi-directional best match)算法寻找模体配对,见图2(c),其中,若一个模体与另外一个数据集中多个模体都最相似,则选取靠前的模体进行配对.
2.2 模体相似多部图的CLIMP聚类
对于配对后的模体相似多部图,运用CLIMP算法[14]进行团(即每对顶点均连接的子图)融合聚类,并形成聚类编号,见图2(d).每个聚类中高度相似的模体分别来自于不同的数据集,这些相似的模体可能是同一转录因子在不同数据集的同一模体.因为同一转录因子可能在多个ChIP数据集中作为辅调控因子或主调控因子出现,因此对应的模体会在多个数据集中被反复识别.
2.3 构建模体共现多部图
对得到的团融合聚类构建模体共现多部图,计算不同聚类中属于相同数据集的每对模体的共现分数.对于数据集Md中的模体Md(i)和Md(j),共现分数Sc为
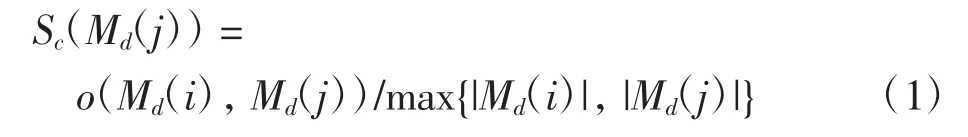
其中:|Md(i)|和|Md(j)|分别为模体Md(i)和Md(j)含有顺式调控元件结合峰的数量;o(Md(i),Md(j))代表这2个模体中都含有的顺式调控元件的结合峰的数量.若共现分数不小于阈值α,则视其为共现模体,将之连接,最终形成模体共现多部图,见图2(e).基于REDfly数据库[15]已有顺式调控模块的训练,阈值α的取值为0.7.
2.4 模体共现多部图的CLIMP聚类
对模体共现多部图进行CLIMP聚类,得到模块类.聚类结果即为顺式调控模块,并按下式由小到大进行排序

其中:M为聚类后的模块;|M|为M中含有模体的数量;m为模块中的模体;i(m)为模体m在团融合聚类后的聚类编号.SM的值越小,则顺式调控模块M就越可能是真实的.将少于2个模体的聚类舍弃.见图2(f).
3 实验结果
结合峰长度分布密度见图3.图中,虚线为结合峰长度分布密度,实线为结合峰长度的累积分布,可见结合峰的大部分长度约为1 000,有0.62%的结合峰长度大于5 000,由于其质量不高,所以不使用这部分数据.由FisherNet查找的模体的信息含量分布密度见图4.由图4可见,162个数据集中的模体(有6个数据集包含模体少于2个,被丢弃)具有较高信息含量.在各个数据集输出的前20个模体中,包含99个已知模体,并且被FisherNet程序优先识别.
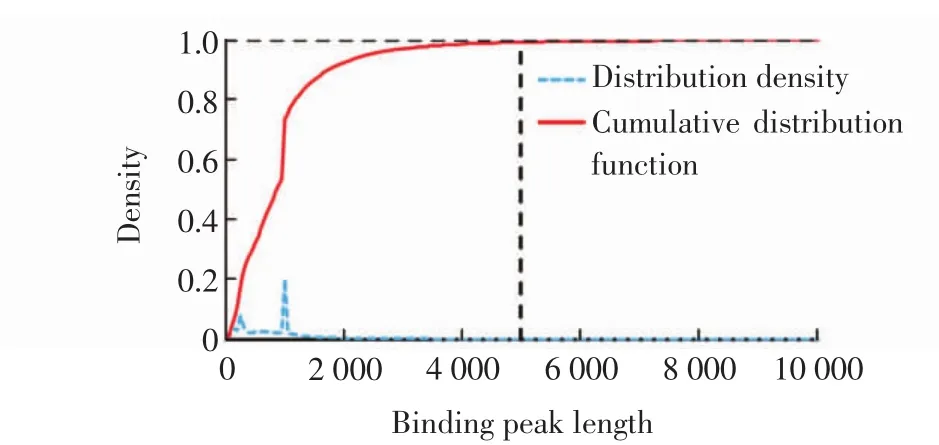
图3 结合峰长度分布密度Fig.3 Distribution density of binding peak length
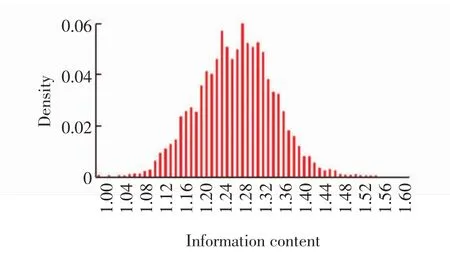
图4 模体信息含量分布密度Fig.4 Distribution density of information content of motifs
将本算法(A)和DePCRM算法(B)应用于162个ChIP数据集,模体和顺式调控模块预测结果见表1.其中,已知顺式调控模块数量为1 330个(REDfly数据库).若一个已知的顺式调控模块与预测的顺式调控模块有至少一半长度是重叠的,则将其视为全覆盖.

表1 本研究算法(A)和DePCRM算法(B)预测结果Tab.1 Predictions of algorithms of this research(A)and DePCRM(B)
由表1可见,在模体发现中,本算法输出每个数据集中最优的模体,得到了3 240个模体,其中包含1 214个已知的顺式调控模块(占已知数量的91.28%);而DePCRM算法由于并未考虑模体的优劣,因此输出模体数量较多,为17890个,其中包含1 061个已知的顺式调控模块(占已知数量的79.77%).在顺式调控模块预测中,本算法得到的1 346个模块中有1 103个已知模块(占已知数量的82.93%);而DePCRM算法得到的115 932个模块中有947个已知模块(占已知数量的71.20%).以上数据说明,本算法在顺式调控模块的预测中较DePCRM有更高的覆盖率和敏感性.
顺式调控模块长度和相邻顺式调控元件间距离分布密度见图 5(a)和(b).由图 5(a)可见,本算法预测的顺式调控模块比已知的顺式调控模块的长度短.由图5(b)可见,预测结果的相邻顺式调控元件间距离与已知的顺式调控元件比较相似,一部分距离比已知的短.这表明可能遗漏了顺式调控模块中的某些顺式调控元件,尤其是两端的,这可能是由于ChIP数据没有足够多样化的信息.
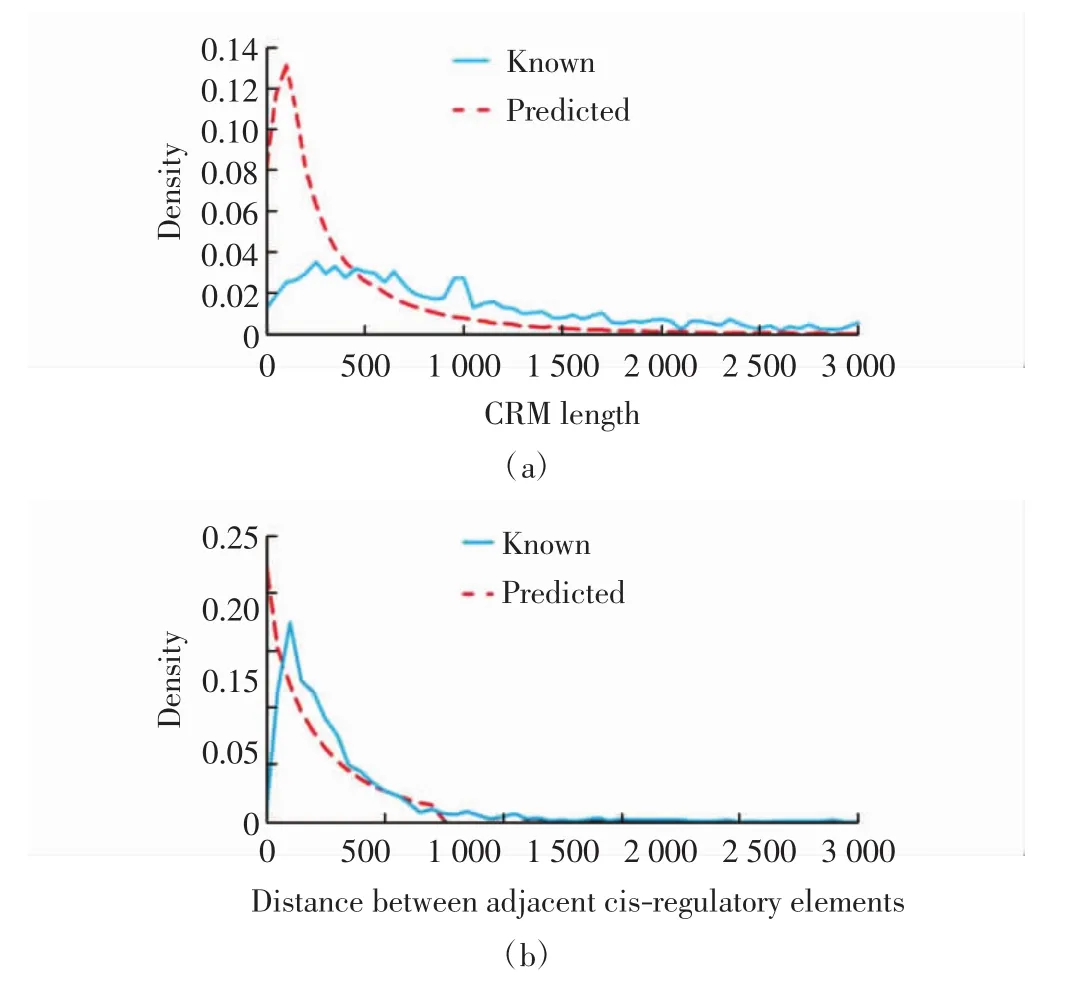
图5 顺式调控模块长度预测结果Fig.5 Prediction results of CRM length
4 结论
本文利用大量的ChIP数据集实现了全基因组范围的顺式调控模块的从头预测.通过识别最优表达的、组合的模体,完成了对顺式调控模块的预测.预测结果覆盖了数据集中已知顺式调控模块的82.93%.这些预测的顺式调控模块比随机选择的序列更保守,更有可能具有调控功能.
与已有的DePCRM算法相比,本文采用了2个多部图和2次CLIMP聚类,比DePCRM算法更简便快速.本算法不采用共现对的概念,克服了模体以偶数对出现的缺点.当有足够多数量的、不同种类的其他真核生物ChIP数据集时,本算法可推广到该类真核生物,用来预测其顺式调控模块.
参考文献:
[1]PEPKE S,WOLD B,MORTAZAVI A.Computation for ChIP-seq and RNA-seq studies[J].Nature Methods,2009,6(11):22-32.
[2]PARK P J.ChIP-seq:Advantages and challenges of a maturing technology[J].Nature Reviews Genetics,2009,10(10):669-680.
[3]HAWKINS R D,HON G C,REN B.Next-generation genomics:An integrativeapproach[J].NatureReviewsGenetics,2010,11(7):476-486.
[4]LAIRD P W.Principles and challenges of genome-wide DNA methylation analysis[J].Nature Reviews Genetics,2010,11(3):191-203.
[5]MASTON G A,EVANS S K,GREEN M R.Transcriptional regulatory elements in the human genome[J].Annual Review of Genomics and Human Genetics,2006,7(1):29-59.
[6]NEGRE N,BROWN C D,MA L J,et al.A cis-regulatory map of the drosophila genome[J].Nature,2011,471(7339):527-531.
[7]GERSTEIN M B,LU Z J,NOSTRAND E L V,et al.Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project[J].Science,2010,330(6012):1775-1786.
[8]MENG N,TABARI E S,SU Z C.De novo prediction of cis-regulatory elements and modules through integrative analysis of a large number of ChIP datasets[J].BMC Genomics,2014,15(1):1047-1066.
[9]CONSORTIUM T M,ROY S,ERNST J,et al.Identification of functional elements and regulatory circuits by Drosophila modENCODE[J].Science,2010,330(6012):1787-1797.
[10]LI X Y,MACARTHUR S,BOURGON R,et al.Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm[J].Plos Biology,2008,6(2):365-388.
[11]ZHANG Y,LIU T,MEYER C A,et al.Model-based analysis of ChIP-seq(MACS)[J].Genome Biology,2008,9(9),DOI:10.1186/gb-2008-9-9-r137.
[12]张志红.基于ChIP-seq数据集的顺式调控模块发现算法研究[D].天津:天津师范大学,2017.ZHANG Z H.Algorithm for Finding Cis-Regulatory Module Based on ChIP-seq Datasets[D].Tianjin:Tianjin Normal University,2017(in Chinese).
[13]ZHANG S Q,ZHOU X,et al.SPIC:A novel similarity metric for comparing transcription factor binding site motifs based on information contents[J].BMC Systems Biology,2013,7(2):1-8.
[14]ZHANG S Q,CHEN Y.CLIMP:Clustering motifs via maximal cliques with parallel computing design[J].Plos One,2016,11(8):1-17.
[15]IVAN A,HALFON M S,SINHA S.Computational discovery of cisregulatory modules in Drosophila,without prior knowledge of motifs[J].Genome Biology,2008,9(1):1-17.
