三种7-乙酰氨基-1,8-萘啶衍生物荧光探针的理论光谱
2017-10-13周俊平迟绍明云南师范大学化学化工学院云南昆明650500
周俊平 迟绍明(云南师范大学化学化工学院, 云南 昆明 650500)
标准与检测
三种7-乙酰氨基-1,8-萘啶衍生物荧光探针的理论光谱
周俊平 迟绍明(云南师范大学化学化工学院, 云南 昆明 650500)
采用量子力学的密度泛函B3LYP/6-31G(d)方法,研究含有不同取代基的三种7-乙酰氨基-1,8-萘啶衍生物分子的结构与理论光谱。分别在气相和液相中,对三种7-乙酰氨基-1,8-萘啶衍生物分子进行结构优化,并计算其紫外可见吸收光谱。不管在气相还是在液相,三种衍生物HOMO和LOMO能极差ΔE均很小。并且与气相的B3LYP/6-31G(d)计算所得到的结果相比较,液相B3LYP/6-31G(d)计算得到的的三种衍生物的最大吸收波长全部都产生了不同程度的红移。
1,8-萘啶衍生物;理论光谱;吸收光谱;TD-DFT
Abstract:The structure and theoretical spectra of three kinds of 7- acetyl -1,8- naphthalene derivatives with different substituents were studied by quantummechanical density functional B3LYP/6-31G(d) method.In this paper,the structures of three kinds of 7- acetyl -1,8-naphthalene derivatives were optimized in the gas and liquid phases,and the UV Vis spectra were calculated.No matter in the gas phase and in liquid phase,three derivatives of HOMO and LOMO to the E range are very small.Compared with the results of B3LYP/6-31G(d)calculation,the maximumabsorption wavelength of the three derivatives obtained by liquid phase B3LYP/6-31G(d) has produced different degrees of red shift.
Key Words:1,8- naphthalene derivative; Theoretical spectrum; absorption spectrum; TD-DFT
1 引言
1.1 荧光分子探针
我们需要得到的有些信息是不能被直接获取的,所以我们常常要用间接的手段来获取。我们可以将它们转化可测量的荧光信号。荧光分子探针就是起到这个作用。它可以在紫外-可见-近红外区可以产生特征荧光,方法简便,在很多方面都适用。灵敏度极强、选择性极好以及简单便捷都是荧光探针分子的特点,并可长距离的发光而不受外界磁场的干涉,受到科研工作者青睐。荧光分子的必备条件一是具有长共轭结构。荧光分子的必要条件二是分子的刚性。当拥有前者即长共轭结构时,荧光强度随电子共轭的水平增强而增强;同理,具有后者即分子的刚性时,同样的共轭程度下荧光强度也会随着刚性的增强而增大。大分子的吸光截面受其结构的影响,其平面结构可以有力的增大其吸光截面,从而提高摩尔吸光系数,使得荧光强度也随着增大。众多研究表明,化合物想要获得较高的荧光性能必要条件就是化合物应当具有刚性结构特别是平面结构。若想要提高大分子内电子的流动性,就要想办法提高分子的共平面性,只要刚性增加,以上过程就能够实现;与此同时也能降低由于震动引起的内转化几率,都对产生荧光有促进作用。荧光强度与摩尔吸光系数以及大分子的吸光截面都有关系,如平面结构的吸光截面较大,其摩尔吸光系数和荧光强度也明显大于其他结构。
1.2 1,8-萘啶衍生物
1,8-萘啶衍生物的两个配位氮原子间的距离为0.2307nm[1,2]。能够符合刚性配位的条件,拥有共轭结构,即发光材料的特征结构。再加上他们具有刚性平面氮杂环结构,使1,8-萘啶衍生物具有独特的特点:广谱的金属螯合能力、独特的荧光性质、优良的生物相容性等[3,4]。由于同时拥有优良的光化学、光物理性质和特殊的药理活性,1,8-萘啶衍生物类化合物在将来相干领域中的运用前景看好。该类化合物不仅分子体积小,而且发光量子效率高,对环境更是敏感,基于这些性质可用于制造生物荧光分子探针[3]。识别研究材料中的生物分子和金属离子是荧光探针的主要作用,探针在诸多领域中有广泛使用也主要得益于其显著的光致发光特性以及因为快速应对环境条件表现出来的光谱变化。1,8-萘啶是萘啶化合物中研究最为广泛的一种[5,6]。1,8-萘啶化合物在辨别致病基因及检测基因突变等方面已经表现出优良的运用前景[7]。目前,对1,8-萘啶衍生物的研究仍在持续,对其结构和作用的探索仍是比较新颖的课题。因为对其作为荧光探针传感器的相关研究鲜见,因此对1,8-萘啶类化合物进行研究、合成具有辨别能力的荧光传感器会是一个很有意义的研究方向。李占先等人[8]合成了三种7-乙酰氨基-1,8-萘啶衍生物。他们对其进行了性质表征,然而他们并没有给出这些化合物的几何结构和详细的光谱信息。本文采用密度泛函理论优化了这三种7-乙酰氨基-1,8-萘啶衍生物的几何构型,接着在B3LYP/6-31G(d)水平下计算了这些配合物的紫外可见吸收光谱。
2 计算方法
整个计算过程主要分为气相中的结构优化、气相中吸收光谱计算、液相中的结构优化、液相中吸收光谱计算四个部分进行。用Gaussian View 5.0得到三种化合物结构图。本研究所考察的三种7-乙酰氨基-1,8-萘啶衍生物荧光探针分子的结构如图1所示,分别命名为A,B,C。接着选用Gaussian程序包中的B3LYP/6-31G(d)的办法分别在气相和液相优化三种7-乙酰氨基-1,8-萘啶衍生物分子基态结构。在把优化结构作为前提的基础下,采用B3LYP/6-31G(d)和含时密度泛函(TD-DFT)的办法计算气相和液相中三种7-乙酰氨基-1,8-萘啶衍生物的紫外可见吸收光谱。将结构优化得到的结果在Gaussian View 5.0中打开,可查看键长、键角和二面角等结构参数以及前线分子轨道能量;而气相和液相中的光谱,将计算得到的结果用SWizard程序处理导出后,可得到紫外光谱图。
3 结果与讨论
3.1 基态几何构型
研究的7-乙酰氨基-1,8-萘啶衍生物分子结构和原子编号如图一所示,氢原子未列入。
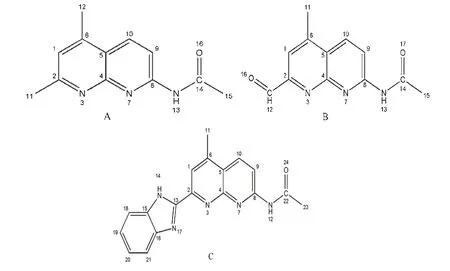
图1 三种不同取代基的7-乙酰氨基-1,8-萘啶衍生物荧光探针分子的结构式
3.2 基态几何结构
三种7-乙酰氨基-1,8-萘啶衍生物分子的结构参数列于表1中。三种衍生物C-C键键角均约为120度,因为碳原子是SP2杂化。对于化合物A,除乙酰氨基外,二面角接近0度或者180度,说明1,8-萘啶和甲基上的碳原子在一个平面上。对于化合物B,除乙酰氨基外,二面角接近0度或者180度,说明1,8-萘啶、甲基上的碳原子和醛基都在一个平面上。对于化合物C,除乙酰氨基外,二面角接近0度或者180度,说明1,8-萘啶、甲基上的碳原子和苯并咪唑都在一个平面上。除乙酰氨基和甲基上的氢外,其他原子则均在一个平面上,分子共平面程度大。溶剂对化合物结构参数影响很小,几乎没影响。
3.3 前线分子轨道分析
将计算结果在Gaussian View 5.0中打开后,可查看其化合物的前线分子轨道能量。前线分子轨道是由分子中能量最高的占有轨道HOMO和能量最低的空轨道LUMO共同构成的。分子的前线区域轨道能量,特别是HOMO和LOMO之间的能极差ΔE是反映物质导电性质和发光性质的一个重要参数[9,10]。下表4列出了三种7-乙酰氨基-1,8-萘啶衍生物分别在气相和液相CH2Cl2中,由高斯G09计算得到的的前线轨道能量。三种衍生物均有共轭体系存在。三种衍生物HOMO和LOMO能极差ΔE均很小。在气相中化合物A、B和C的ΔE分别为0.17571au,0.14965au和0.14908au。在液相CH2Cl2中 化 合 物A、B和C的ΔE分 别 为0.17947au,0.13905au和0.14665au。说明溶剂对前线轨道能量有一定影响的。
3.4 紫外吸收光谱
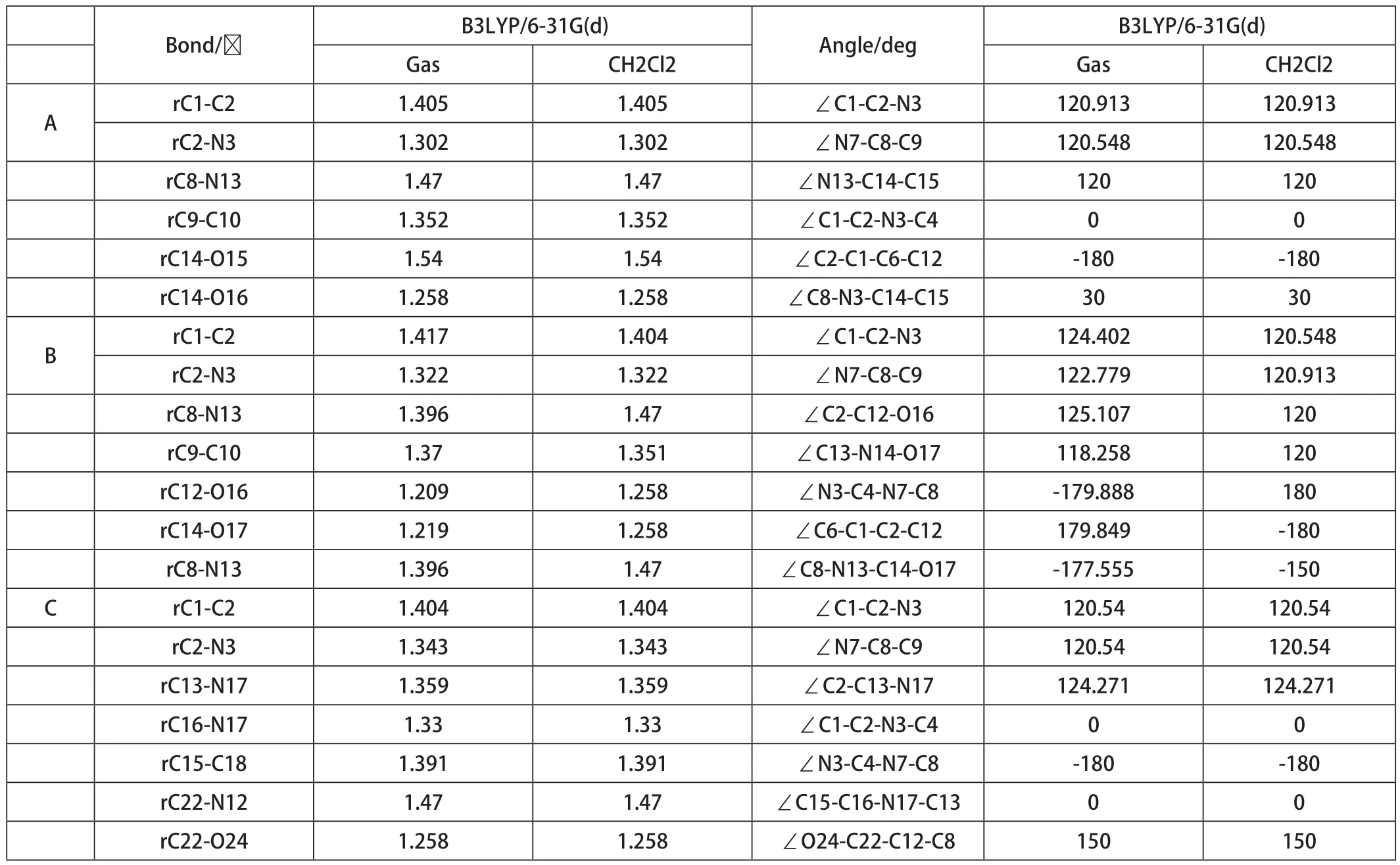
表1 三种卤化BODIPY染料分子的理论几何参数
将计算得到的结果用SWizard程序处理后,用Origin进行画图并进行归一化处理。如图2是三种7-乙酰氨基-1,8-萘啶衍生物分别在气相和液相CH2Cl2中的理论光谱。由图可以看出,在气相和液相CH2Cl2中测定化合物A、B和C的紫外一可见吸收光谱,同种物质在气相和液相中的吸收光谱变化不大,且3个化合物的吸收光谱在不同的存在状态下有相同的变化趋势。A,B,C在250nm和320nm附近有吸收。A、B在210-250nm,250-300nm 范围有吸收,因为含有不饱和酮基和苯环。C在250-300nm,300nm以上有吸收,因为有多个苯环,有较大的共轭体系。在气相中A,B和C的最大吸收波长分别为283.35nm,249.42nm和338.44nm。在液相CH2Cl2中化合物A、B和C的最大吸收波长分别283.41nm,307.48nm,348.12nm。 与 气 相 的B3LYP/6-31G(d)计算结果相比,液相B3LYP/6-31G(d)计算的三种衍生物的最大吸收波长均发生了不同程度的红移。因为存在溶剂效应。
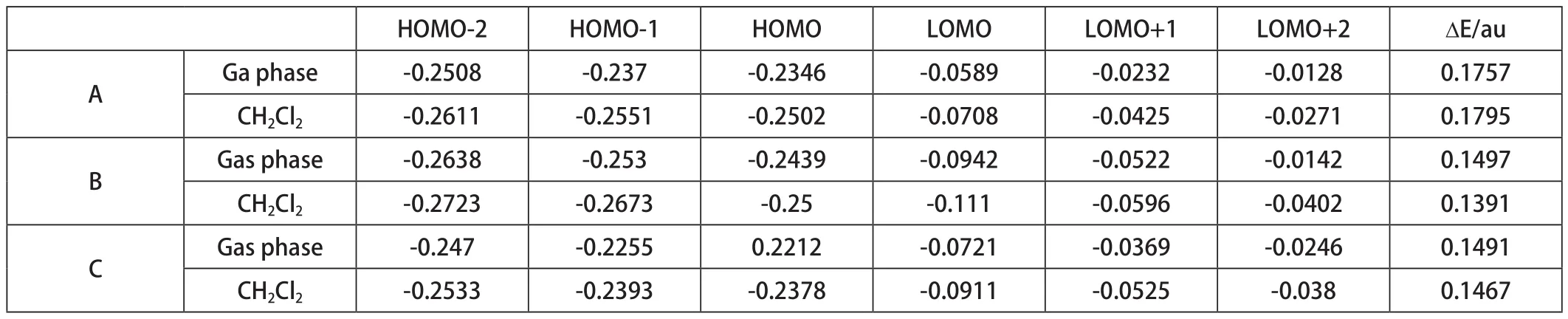
表2 三种衍生物的能隙和前线分子轨道的能量
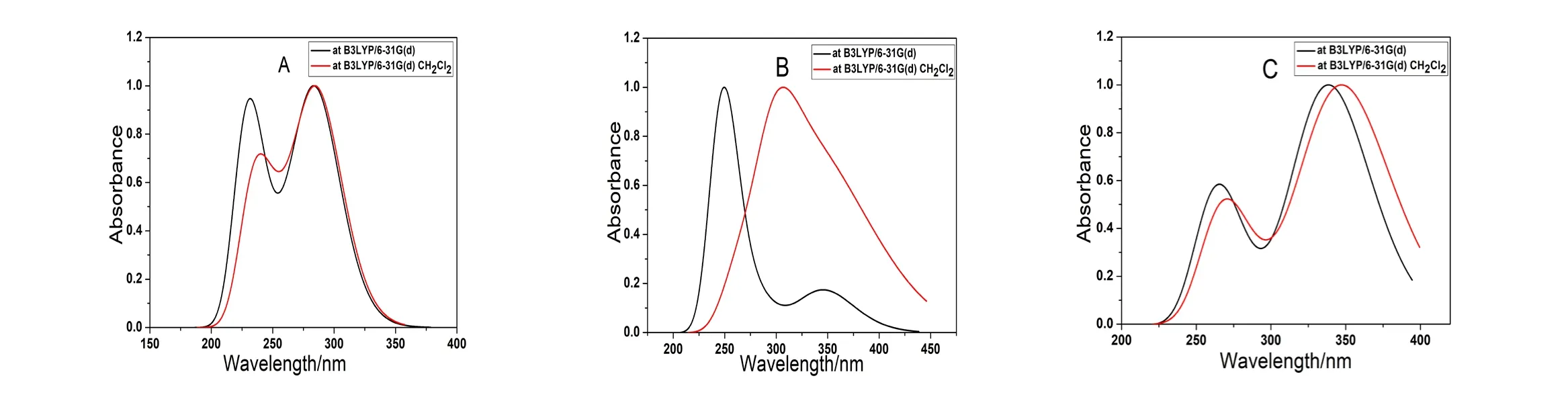
图2 三种7-乙酰氨基-1,8-萘啶衍生物分别在气相和液相CH2Cl2中的理论光谱
4 结语
用量子力学的密度泛函B3LYP/6-31G(d)方法得到三种7-乙酰氨基-1,8-萘啶衍生物基态的几何构型。三种衍生物键角均约为120度,因为碳原子是sp2杂化。sp2杂化的键角约为120度。除乙酰氨基和甲基上的氢外,其他原则均在一个平面上,分子共平面程度大。溶剂对化合物结构参数影响很小,几乎没影响。探索得到了三种7-乙酰氨基-1,8-萘啶衍生物分子分别在气相和液相中的分子轨道能量和紫外可见吸收光谱图。我们可以从研究结果断然看出,不管在气相还是在液相,三种衍生物HOMO和LOMO能极差ΔE均很小。化合物A、B在210-250nm,250-300nm范围有吸收,因为含有不饱和酮基和苯环。化合物C在250-300nm,300nm以上有吸收,因为有多个苯环,有较大的共轭体系。并且因为存在溶剂效应,与气相的B3LYP/6-31G(d)计算得到的结果相比较,液相B3LYP/6-31G(d)计算得到的三种衍生物的最大吸收波长均发生了不同程度的红移。
[1]冯媛,傅文甫.1,8-萘啶衍生物的结构、光物理性质及其应用[J].影像科学与光化学,2013,(04)∶241-256.
[2]陈勇.1,8-萘啶衍生物及其d~(10)金属配合物∶合成、结构及光物理性质[D].中国科学院研究生院(理化技术研究所),2007.
[3]石彩霞,赵万莹,刘兴江,等.1,8-萘啶衍生物研究进展[EB/OL].北京∶中国科技论文在线.2013.8.20.
[4]刘兴江.基于1,8-萘啶和8-羟基喹啉衍生物分子探针的设计合成及其应用[D].郑州大学,2014.
[5]苟高章,吴娜,石玲,等.2-甲基-1,8-萘啶衍生物的甲基溴代反应[J].应用化学,2014,(11)∶1268-1272.
[6]汪帮忠.1,8-萘啶及吡啶类氟硼化合物的合成、光谱性质及密度泛函理论研究[D].云南师范大学,2016.
[7]贾临芳,傅文甫.发光1,8-萘啶衍生物及其铜(Ⅰ)配合物的合成及光性能研究[J].感光科学与光化学 ,2005,04∶316.
[8]Z.X.Li,W.Y Zhao,X.Y.Li,et al.1,8-Naphthyridine-Derived Ni2+/Cu2+-Selective Fluorescent Chemosensor with Different Charge Transfer Processses.Inorganic Chemistry,2012.51(22)∶p.12444-12449.
[9]迟绍明,李立,陈勇,等.1,8-萘啶衍生物分子结构与理论电子光谱[J].光谱学与光谱分析,2010,(03)∶586-590.
[10]范建训,仁爱民,薄冬生等.7-氮杂吲哚衍生物分子基态和激发态性质的理论研究[J].高等学校化学学报 ,2006,27(6)∶1091.
周俊平(1992-),女,汉,大学本科,研究生在读,理学学士。
基金名称(基金号) 迟绍明-新型1,8萘啶衍生物及Cu(1)配合物设计、合成及光谱的理论和实验(21262049)
