亚硝酸还原酶基因克隆、表达与纯化
2017-08-09魏计东张庆芳窦少华迟乃玉王晓辉大连大学生命科学与技术学院辽宁大连116622辽宁省海洋微生物工程技术研究中心辽宁大连116622
魏计东,张庆芳,窦少华,于 爽,迟乃玉,王晓辉,* (1.大连大学生命科学与技术学院,辽宁大连 116622; 2.辽宁省海洋微生物工程技术研究中心,辽宁大连 116622)
亚硝酸还原酶基因克隆、表达与纯化
魏计东1,2,张庆芳1,2,窦少华1,2,于 爽1,2,迟乃玉1,2,王晓辉1,2,*
(1.大连大学生命科学与技术学院,辽宁大连 116622; 2.辽宁省海洋微生物工程技术研究中心,辽宁大连 116622)
该研究通过聚合酶链反应(PCR)方法从木糖氧化产碱菌(AchromobacterxylosoxidansDL-1)基因组DNA中成功克隆含铜亚硝酸还原酶基因。PCR测序表明该基因全长1083个核苷酸,编码360个氨基酸,预测其理论分子质量约38.924 kDa,等电点(pI)4.83,命名为CuNiR(Genbank登录号KX674378)。结构域分析该蛋白编码区包含信号肽,1个铜离子结合位点,1个氧化还原酶催化域。将CuNiR基因构建到pET22b载体,并转化至大肠杆菌(Escherichiacoli,E.coli)BL21(DE3)中诱导表达,十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)检测显示目的蛋白可溶表达。利用镍柱亲和层析纯化重组蛋白,酶活检测显示比活力为123.82 U/mg,为后期含铜亚硝酸还原酶(CuNiR)的生化性质表征奠定理论基础。
木糖氧化产碱菌DL-1,含铜亚硝酸还原酶(CuNiR),克隆,表达,纯化
自然界中氮素循环是各个元素循环的中心,几乎所有有机体都需要有机氮作为细胞的必需成分,氮元素成为地球表面生物量增长的限制性因素[1]。在氮素循环中,固氮作用与脱氮作用是两个十分重要的环节,生物圈中氮素的引入主要是通过固氮作用,而最终实现对大气中蕴藏丰富的氮素的利用。与此同时,生物圈中氮素的释放则离不开脱氮作用。脱氮作用是指通过一系列的厌氧电子传递反应把硝酸经由亚硝酸、一氧化氮、氧化亚氮等中间体还原为氮气或氨的过程[2]。反硝化作用主要由反硝化细菌[3]在微氧或厌氧条件下完成。
亚硝酸还原酶是催化亚硝酸盐还原为一氧化氮(NO)的酶。反硝化途径[4]由一系列的还原反应组成,每一步都有特定的酶来催化完成。而亚硝酸还原酶最终实现了将溶液态的亚硝酸向气态NO的相变,直接导致了氮素从陆表环境中的流失,可以认为是反硝化途径中最为关键的一步[5];其次,利用亚硝酸还原酶降解发酵食品中的亚硝酸盐是应对亚硝酸盐污染的根本对策。对许多细菌中的亚硝酸还原酶分析表明它有两种类型:以细胞色素cd1(cd1NiR)和金属Cu(CuNiR)为辅基,其中细胞色素cd1NiR更为经常的发生[6]。两种类型的亚硝酸还原酶在结构和氧化还原中心的差异很大,但催化相同的单电子亚硝酸还原反应。第一个含铜亚硝酸还原酶是在1955年,由Iwasaki[7]等人于自名古屋大学的土壤中分离的Alcaligenes(现在称为Achromobacter)xylosoxidans NCIB11015菌株中发现的。亚硝酸还原酶中CuNiR含量相对较少,但是表现了更广泛的环境、地域、种群等分布上的多样性。迄今为止,已在十几种具有反硝化作用的微生物中明确鉴定出CuNiR的存在[8]。最新研究热点主要集中于亚硝酸还原酶催化作用的分子机理,具体涉及电子在两个类铜位点之间的传递,以及CuNiR结合于类型2铜位点(T2Cu)的亚硝酸还原反应发生情况[9];而关于该酶的基因水平研究较少[10]。
表1 PCR引物序列Table 1 PCR primer sequences
本文在分子水平上实现编码含铜亚硝酸还原酶基因的克隆、表达及纯化。含铜亚硝酸还原酶基因CuNiR的克隆、表达以及酶的纯化是重组基因工程菌生产亚硝酸还原酶(NiR)产业化应用的重要研究基础。通过对CuNiR基因克隆和表达的研究,有助于我们了解亚硝酸还原酶(NiR)基因结构与表达产物之间的关系,并为根据不同应用目的进行亚硝酸还原酶(NiR)分子工程化的改造奠定基础。
1 材料与方法
1.1 材料与仪器
木糖氧化产碱菌(AchromobacterxylosoxidansDL-1) 由本实验室分离并保存;大肠埃希氏菌(Escherichiacoli,E.coli)JM109和BL21(DE3) 为本实验室保存,分别为基因克隆和表达宿主菌;表达载体pET-22b(+) 购自Novagen公司;引物合成和基因测序 由宝生物工程(大连)有限公司完成;十二烷基硫酸钠(SDS)、琼脂糖、Tris Promega公司;LA Taq DNA聚合酶、DNA凝胶回收试剂盒、质粒纯化试剂盒、低分子量蛋白Marker 宝生物(大连)工程有限公司;蛋白浓度测定试剂盒 碧云天公司;LB培养基(1000 mL):胰蛋白胨10 g,酵母提取物 5 g,NaCl 10 g,pH7.0;LB固体培养基需向液体培养基内加入2%(w/v)琼脂;LA培养基:LB液体或固体培养基中加入终浓度100 μg/mL氨苄青霉素。
PCR扩增仪 TaKaRa公司;GS-158低温台式离心机 BECKMAN公司;J21-M高速冷冻离心机 BECKMAN公司;台式冷冻离心机 Eppendorf公司;CH1015超级恒温水浴槽 上海恒平仪器厂;Inazge MlasterRVDS电泳成像系统 Parmacia Biotech公司;DYY-III形电泳槽 北京六一仪器厂;Milli-Q Academic超纯水器;pH计 BECKMAN公司。
1.2 实验方法
1.2.1CuNiR基因的克隆与序列分析 从平板上挑取菌株AchromobacterxylosoxidansDL-1单菌落,接种于50 mL LB液体培养基,37 ℃、160 r/min振荡培养12 h,12000 r/min离心10 min收集菌体,按细菌基因组DNA提取试剂盒操作步骤提取基因组DNA。
以基因组DNA为模板,根据Kataoka K等[11]的方法设计含铜亚硝酸还原酶基因序列(AB013078)的两对引物,见表1。PCR反应条件为:94 ℃ 2 min,1个循环;94 ℃ 30 s,55 ℃ 45 s,68 ℃ 7 min,30个循环;68 ℃ 15 min,1个循环。PCR扩增产物经1%(w/v)琼脂糖电泳检测,将PCR产物大小正确的克隆送宝生物工程(大连)有限公司进行测序。采用Vector NTI Suite软件进行开放阅读框(open reading frames,ORF)分析。通过NCBI上的本地序列基本搜索工具(basic local alignment search tool,BLAST)分析序列同源性。采用在线的简单模块构架搜索工具(simple modular architecture research tool,SMART)进行序列结构域分析。
1.2.2CuNiR基因的表达与纯化 按实验方法构建CuNiR基因的表达载体,热转化至E.coliCompetent Cell JM109中,涂布平板培养菌体。随机挑取的12个单菌落进行PCR鉴定,鉴定正确的质粒分别命名为CuNiR-P-1和CuNiR-P-2,1%琼脂糖凝胶电泳检测。CuNiR基因(去除信号肽)和原核表达载体pET22b(+)的PCR产物经NdeI/HindⅢ双酶切后用T4 DNA 连接酶连接,构建重组原核表达载体pET22b-CuNiR,并转化至大肠杆菌感受态细胞E.coliBL21(DE3)中。双酶切鉴定正确的阳性克隆接入含氨苄的LB培养基中,37 ℃培养12 h。按1%的接种量接种到含氨苄的LB培养基中。当OD600 nm达到0.6~0.8左右,加入1.0 mmol/L的IPTG,37 ℃,100 r/min诱导2 h。4 ℃,6000×g离心10 min,收集菌体,用适量PBS缓冲液(pH7.0)洗涤菌体并重悬。离心管置于冰上超声(400 W)破碎三次,每次10 s,间隔1 min,直至菌体透明。4 ℃,8000×g离心20 min,收集上清液,即为粗蛋白。粗蛋白用Ni2+-NTA柱(Novagen)进行亲和层析纯化[12],洗脱的蛋白液4 ℃保存。蛋白样品经变性聚丙烯酰胺凝胶电泳(SDS-PAGE)检测,其中浓缩胶浓度5%,分离胶浓度12%,用考马斯亮蓝R-250染色。低分子量蛋白Marker用于SDS-PAGE。
1.2.3 蛋白质浓度测定 蛋白浓度通过Bradford蛋白浓度测定试剂盒(Bradford Protein Assay Kit)测定。
1.2.4CuNiR酶活性的测定CuNiR活性测定采用Na2S2O4-MV法[13]。测定酶活反应体系250 μL:0.1 mol/L磷酸盐缓冲液(pH7.5)125 μL,0.1 mol/L NaNO212.5 μL,0.1 mol/L MV 7.5 μL,0.1 mol/L Na2S2O440 μL,酶液50 μL。30 ℃水浴中反应10 min,剧烈振荡终止反应(以磷酸缓冲液为空白)。取10 μL加格利斯试剂显色,在波长540 nm处比色法测定亚硝酸盐的变化。
硝酸还原酶活力单位通过在25 ℃下,每分钟还原1 μmol亚硝酸盐所消耗的酶量来表示。比活力用1 mg蛋白质中酶的活力单位数来表示。
2 结果与分析
2.1CuNiR基因的扩增与序列分析
以AchromobacterxylosoxidansDL-1基因组DNA为模板,PCR扩增得到一条长度约1083 bp的DNA片段(图1)。DNA测序结果表明:该基因包含1083个核苷酸构成的开放阅读框架,命名为CuNiR,该基因编码360个氨基酸,理论分子量38.924 kDa,等电点为4.83。本文首次报道从AchromobacterxylosoxidansDL-1中克隆亚硝酸还原酶基因。Blast比对分析该序列同已发表的A.xylosoxidansAU1011亚硝酸还原酶(GeneBank No. AB013078)氨基酸同源性达96%。但未见比对菌株中该蛋白表达纯化研究。结构域分析该基因包含一段24个氨基酸构成的信号肽,1个铜离子结合结构域和1个氧化还原酶催化域。

图1 含铜亚硝酸还原酶基因的PCR扩增Fig.1 Agarose gel electrophoresis of the CuNiR gene through PCR注:M:DNA Marker DL2000;1:CuNiR gene。
2.2CuNiR基因测序载体的鉴定
将以CuNiR-F2/R2为引物PCR扩增得到的目的片断CuNiR基因克隆至pMD19-T simple载体中,构建了重组质粒pMD19-T simple/CuNiR,通过1%琼脂糖凝胶电泳检测。由图2可见在约3.7 kbp处得到4条明亮清晰DNA条带,其中去除信号肽CuNiR基因片段大小约1011 bp,pMD19-T simple载体大小为2692 bp,结果显示目的片断已经成功克隆至质粒载体中,成功构建重组质粒pMD19-T simple/CuNiR,进一步测序验证正确的目的基因进行后续分析。

图2 重组质粒pMD19-T simple/CuNiR的PCR电泳Fig.2 Agarose gel electrophoresis of recombinant plasmid pMD19-T simple/CuNiR注:M:Lambda DNA/Hind III Marker; 1~4:重组质粒pMD19-T simple/CuNiR。
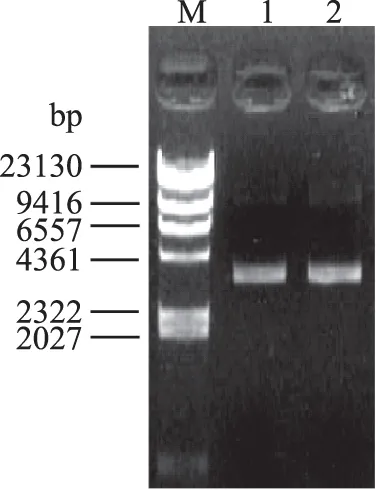
图3 重组质粒pET-22b-CuNiR琼脂糖凝胶电泳Fig.3 Agarose gel electrophoresis of pET-22b/CuNiR plasmid注:M:λ-Hind Ⅲ DNA Marker;1:CuNiR-P-1;2:CuNiR-P-2。
2.3CuNiR基因表达载体的构建与鉴定
提取重组表达质粒CuNiR-P-1和CuNiR-P-2进行1%琼脂糖凝胶电泳检测。结果如图3所示,重组表达质粒与预期大小一致。
将质粒CuNiR-P-1和CuNiR-P-2分别用限制性内切酶NdeⅠ/HindⅢ进行双酶切,取20 μL酶解产物上样进行1%琼脂糖凝胶电泳检测,结果如图4所示。从图4中可以看出,两个重组质粒酶解产物片段大小分别约为1 kbp和5.3 kbp左右,说明目的CuNiR基因已成功的亚克隆至表达载体pET-22b(+)中。
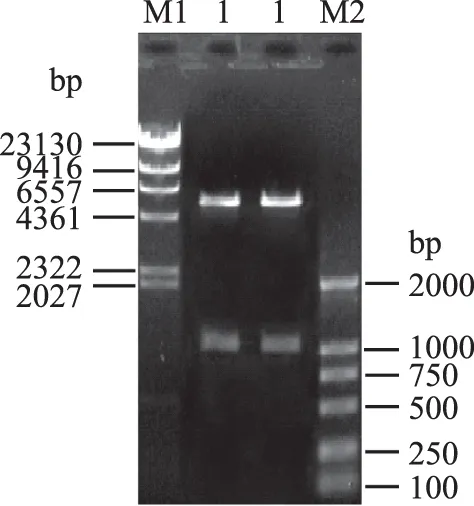
图4 质粒CuNiR-P-1和CuNiR-P-2酶切产物回收Fig. 4 Agarose gel electrophoresis of recombinant expression plasmid pET-22b(+)/CuNiR digested with restriction enzyme注:M1:λ-Hind Ⅲ DNA Marker; 1:CuNiR-P-1 Nde I/HindⅢ酶切; 2:CuNiR-P-2 Nde I/HindⅢ酶切; M2:DNA Marker DL2000。
2.4CuNiR目的蛋白诱导表达与检测
将经鉴定正确的原核重组表达质粒载体pET-22b(+)/CuNiR(CuNiR-P-1)转化至表达宿主菌E.coliBL21(DE3)中诱导表达。菌体裂解物进行SDS-PAGE电泳,由图5可知,泳道1~3空表达载体pET-22b(+)宿主菌BL21(DE3)未见目的蛋白,而泳道4和5约36 kDa位置处出现特异条带,与预期表达的相对分子质量相符,说明目的蛋白CuNiR在宿主菌E.coliBL21(DE3)中成功表达,且在全细胞和细胞浆的可溶性蛋白中检测到目的蛋白。由此可知,目的蛋白CuNiR在大肠杆菌中可溶表达。
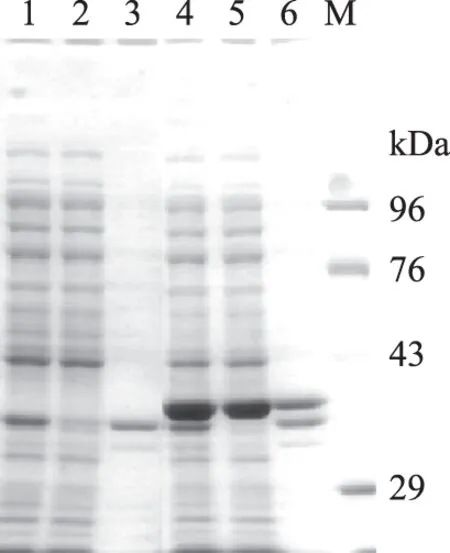
图5 目的蛋白CuNiR的SDS-PAGE检测结果Fig.5 SDS-PAGE analysis of CuNiR protein注:1:pET-22b(+)全细胞;2:pET-22b(+)上清; 3:pET-22b(+)沉淀;4:pET-22b(+)/CuNiR全细胞; 5:pET-22b(+)/CuNiR上清;6:pET-22b(+)/CuNiR沉淀; M:TakaRa Protein Marker(Low)。
2.5CuNiR的纯化与酶活检测
在摇瓶水平,1 L发酵液经1 mmol/L IPTG诱导2 h离心收集菌体,超声破碎后得到CuNiR上清液蛋白。破碎上清液经Ni-NTA纯化重组蛋白,SDS-PAGE检测洗脱收集液,如图6所示。目标蛋白被120、300 mmol/L咪唑溶液洗下收集,获取纯度较高且浓度较大的目标蛋白。纯化后CuNiR总蛋白含量为96 mg,比活力为123.82 U/mg,总活力为11887.08 U。
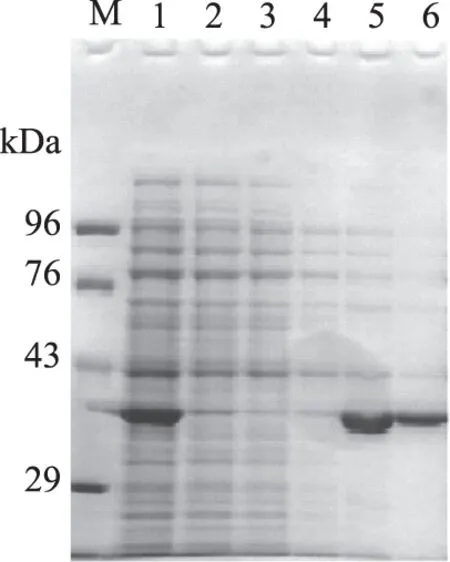
图6 重组含铜亚硝酸还原酶CuNiR的分离纯化Fig.6 Purification of the recomninant CuNiR注:M:低分子量蛋白Marker; 1:E.coli BL21(DE3)/pET22b-CuNiR细胞裂解液; 2:Ni-NTA纯化后CuNiR过柱后洗脱液; 3:磷酸缓冲液(pH7.5)洗脱液;4:20 mmol/L咪唑洗脱液; 5:120 mmol/L咪唑洗脱液;6:300 mmol/L咪唑洗脱液。
3 结论
亚硝酸还原酶大多数为胞内酶,受分离纯化技术的影响,该酶的应用受到了极大限制[14]。本研究所用的pET系列表达载体是有史以来在E.coli中克隆表达重组蛋白功能最强大的系统,保证外源目的基因成功表达与纯化。本实验中我们选择了带有NdeⅠ酶切位点的表达载体pET-22b(+),该表达载体多克隆位点C端有一个能编码6个组氨酸的融合标签His-Tag,6×His通常不影响表达产物的生物学活性,因而不必通过酶水解获得目的蛋白,使表达的整个过程更为简单,同时该融合标签可用于目的蛋白的检测和纯化。在融合标签His-Tag后还相连一个终止密码子TGA,它保证了目的蛋白和His-Tag的完整表达[15-16]。截至目前文献报道亚硝酸还原酶主要集中于甜菜、小麦、菠菜等植物中[17-18],而微生物亚硝酸还原酶报道较少[19-20]。本研究将含铜亚硝酸还原酶基因成功克隆并构建表达载体pET22b,再转化至大肠杆菌(Escherichiacoli,E.coli)BL21(DE3)中诱导表达,目的蛋白经SDS-PAGE检测显示为可溶表达,随后利用镍柱亲和层析纯化蛋白,酶比活可达123.82 U/mg,为下一步酶学性质研究及工业化生产奠定基础。
[1]Orellana L H,Rodriguez-R L M,Higgins S,et al. Detecting nitrous oxide reductase(NosZ)genes in soil metagenomes:method development and implications for the nitrogen cycle[J]. Mbio,2014,5(3):191-194.
[2]Allen J W,Barker P D,Daltrop O,et al. Why isn’t ‘standard’ heme good enough for c-type and d1-type cytochromes[J]. Dalton Transactions,2005,21(21):3410-3418.
[3]Sharma S,Szele Z,Schilling R,et al. Influence of freeze-thaw stress on the structure and function of microbial communities and denitrifying populations in soil[J]. Journal of Experimental Medicine,2006,72(3):2148-2154.
[4]Antonyuk S V,Strange R W,Sawers G,et al. Atomic resolution structures of resting-state,substrate-and product-complexed Cu-nitrite reductase provide insight into catalytic mechanism[J]. Proceedings of the National Academy of Sciences,2005,102(34):41-46.
[5]Hallin S,Lindgren P E. PCR Detection of genes encoding nitrite reductase in denitrifying bacteria PCR detection of genes encoding nitrite reductase in denitrifying bacteria[J]. Applied and Environmental Microbiology,1999,65(4):1652-1657.
[6]Tsuneda S,Miyauchi R,Ohno T,et al. Characterization of denitrifying polyphosphate-accumulating organisms in activated sludge based on nitrite reductase gene[J]. Journal of Bioscience and Bioengineering,2005,99(4):403-408.
[7]Yamaguchi K,Deligeer,Nakamura N,et al. Isolation and characterization of two distinct azurins from Alcaligenes xylosoxidans subsp. xylosoxidans NCIB11015 or GIFU1051[J]. Chemistry Letters,1995,89(5):353-354.
[8]Moura I,Moura J J. Structural aspects of denitrifying enzymes[J]. Current Opinion in Chemical Biology,2001,5(2):168-175.
[9]Bykov D,Neese F. Six-electron reduction of nitrite to ammonia by cytochrome c nitrite reductase:insights from density functional theory studies[J]. Inorganic Chemistry,2015,54(19):3-16.
[10]Suzuki M,Hirai T,Arai H,et al. Purification,characterization,and gene cloning of thermophilic cytochrome cd1 nitrite reductase from Hydrogenobacter thermophilus TK-6[J]. Journal of Bioscience & Bioengineering,2006,101(5):391-397.
[11]Kataoka K,Furusawa H,Takagi K,et al. Functional analysis of conserved aspartate and histidine residues located around the type 2 copper site of copper-containing nitrite reductase[J]. Journal of Biochemistry,2000,127(2):345-348.
[12]李美玉,张庆芳,王晓辉. 几丁质结合蛋白基因克隆、表达与纯化[J]. 中国酿造,2015(11):41-46.
[13]Rosa M. Purification and characterisation of a possible assimilatory nitrite reductase from the halophile archaeon Haloferax mediterranei[J]. FEMS Microbiology Letters,2001,196(2):113-118.
[14]Treusch A H,Leininger S,Kletzin A,et al. Novel genes for nitrite reductase and Amo-related proteins indicate a role of uncultivated mesophilic crenarchaeota in nitrogen cycling[J]. Environmental Microbiology,2005,7(12):85-95.
[15]Ai-Jia J I,Ning X B. Construction and expression of Prokaryotic expression Vector pET28a-EGFP[J]. Journal of Microbiology,2011,31(4):69-73.
[16]Yuan Z G,Zhang J P,Chu Y W,et al. Expression of target gene in eukaryotic cells driven by prokaryotic T7 promoter and its RNA polymerase][J]. Chinese Journal of Biotechnology,2005,21(2):182-186.
[17]杜永成,王玉波,范文婷,等. 不同氮素水平对甜菜硝酸还原酶和亚硝酸还原酶活性的影响[J]. 植物营养与肥料学报,2012,18(3):717-723.
[18]Rajasekhar V K,Oelmüller R. Regulation of induction of nitrate reductase and nitrite reductase in higher plants[J]. Physiologia Plantarum,2006,71(4):517-521.
[19]Prudêncio M,Eady R R,Sawers G. The Blue Copper-Containing Nitrite Reductase from Alcaligenes xylosoxidans:Cloning of the nirA Gene and Characterization,of the Recombinant Enzyme[J]. Journal of Bacteriology,1999,181(8):2323-2329.
[20]Ho W H,Ooi B L. Cytoplasmic expression of the Achromobacter xylosoxidans blue copper nitrite reductase in Escherichia coli and characterisation of the recombinant protein[J]. Protein Expression & Purification,2003,32(2):288-292.
Cloning,expression and purification of copper nitrite reductase
WEI Ji-dong1,2,ZHANG Qing-fang1,2,DOU Shao-hua1,2,YU Shuang1,2,CHI Nai-yu1,2,WANG Xiao-hui1,2,*
(1.School of Life Science and Biotechnology,Dalian University,Dalian 116622,China; 2.Liaoning Technology of Marine Microbiological Engineering Research Center,Dalian 116622,China)
In this study,a novel copper nitrite reductase gene was cloned fromAchromobacterxylosoxidansDL-1 by using a PCR protocol. The gene contained a 1083 bp open reading frame encoding a 360 amino acid protein with an estimated molecular mass of 38.924 kDa and isoelectronic point(pI)of 4.83,namedCuNiR(Genebank no. KX674378). Based on domains analysis,CuNiRhad a signal peptide,a copper binding site classified as Cu-oxidase-3,and one nitrite reductase catalytic domain(Cu-oxidase). A recombinant plasmid,pET22b-CuNiRwas constructed and transformed intoE.coliBL21(DE3). The cells were induced by addition of IPTG.CuNiRhad been successfully expressed by the analysis of sodium dodecyl sulphate-polyacrylamide gel electrophoresis(SDS-PAGE). The recombinant protein was purified by Ni-NTA resin,and the specific activity was 123.82 U/mg,which lay the theoretical basis for biochemical characterization of the copper nitrite reductase.
AlcaligenesxylosoxidansDL-1;copper nitrite reductase;cloning;expression;purification
2016-12-14
魏计东(1991-),男,硕士研究生,研究方向:微生物与酶工程,E-mail:weijidongsk@163.com。
*通讯作者:王晓辉(1981-),女,博士,副教授,研究方向:微生物与酶工程,E-mail:wangxiaohui@dlu.edu.cn。
国家自然科学基金项目(31500039)。
TS201.2
A
1002-0306(2017)14-0101-05
10.13386/j.issn1002-0306.2017.14.020
