一种基于位置信息的高效DNA序列挖掘算法
2017-07-10杨静欣毛国君
杨静欣 毛国君
(中央财经大学信息学院 北京 100081)
一种基于位置信息的高效DNA序列挖掘算法
杨静欣 毛国君
(中央财经大学信息学院 北京 100081)
类Apriori算法在产生频繁模式时需要多次扫描数据库,并且产生大量的候选集;FreeSpan和PrefixSpan等基于投影数据库的算法在产生频繁模式时会产生大量的投影数据库,占用很多内存空间,这些都造成了很大的冗余。针对以往序列挖掘算法存在的不足,提出一种高效的序列挖掘算法——基于位置信息的序列挖掘算法PBSMA(Position-Based Sequence Mining Algorithm)。PBSMA算法通过记录频繁子序列的位置信息来减少对数据库的扫描,利用位置信息逐渐扩大频繁模式的长度,并且借鉴关联矩阵的思想和PrefixSpan算法中前缀的概念,深度优先去寻找更长的关键模式。实验结果证明,无论在时间还是空间上,PBSMA算法都比PrefixSpan算法更高效。
序列挖掘 DNA序列 位置信息 关联矩阵 前缀
0 引 言
序列挖掘是数据挖掘中非常重要的一个研究领域,一般是指从序列数据库中发现蕴含在其中的,相对于时间或者其他顺序高频率出现的子序列,并称这个高频子序列为序列模式。序列挖掘的概念最早由Agrawal等在针对超市中购物篮数据的分析中提出[1],目的是发现某一时间段内客户的购买活动规律。序列挖掘经过近20年的发展,其应用范围已经不再局限于交易数据库,在商业,消费者行为分析,股票趋势预测,生物序列分析,Web挖掘等领域都有重要的应用。商业组织利用序列模式去研究客户购买行为模式特征、计算生物学中序列模式挖掘来分析不同氨基酸突变模式、用户Web访问模式预测以及DNA序列分析和谱分析,尤其在DNA分析等尖端科学研究领域、Web访问等新型应用数据源等方面得到了有针对性的研究[2-3]。
DNA序列挖掘是序列挖掘最重要的应用之一,通过挖掘DNA序列,研究者可以发现隐藏在序列数据背后的有价值的规律,用以解读相应生物体中的生命特征。众所周知,生物的遗传信息被储存在DNA中,而DNA是由两条盘绕在双螺旋结构上的线性链组成,每条链可以看成是四种核苷酸A(腺嘌呤)、G(鸟嘌呤)、T(胸腺嘧啶)、C(胞嘧啶)的线性组合,两条线性链严格遵守碱基配对规则,即A与T配对、C与G配对出现。例如,一条DNA序列是
二十一世纪是生物技术的时代,生物技术是当前最热门的研究领域之一[4]。DNA数据量庞大,其中蕴藏着海量的信息,我们有理由相信,DNA序列挖掘将会帮助人类解开更多的基因之谜,在遗传、疾病、物种学上带来更多的突破。DNA全序列具有什么样的结构、由这4个字符排成的看似随机的序列中隐藏着什么规律、各碱基的功能等都有待于进一步的研究。因此,DNA序列模式挖掘成为序列挖掘最重要、最有价值的应用。
1 相关工作
在早期,序列挖掘算法大多基于Agrawal等提出的关联规则算法—Apriori算法[5],该算法的核心理论是频繁模式的任何子模式都是频繁的。Agrawal和Srikant提出的AprioriAll算法、AprioriSome算法和DynamicSome算法都是基于这个思想而提出,成为序列挖掘算法研究的基础[1];1996年,Srikant和Agrawal又提出GSP算法,这是一种广度优先的自下而上的算法,采用不同的剪枝策略产生较少的候选模式,因而效率相对于AprioriAll算法有所提高,是目前引用率较高的算法之一[6];然而类Apriori算法自身存在局限性,学者们纷纷开始寻找更高效的方法。2000年,Han等提出了基于模式增长的FP-growth算法,该算法不产生候选集,而是使用FP-tree的树状结构压缩数据库,然后再利用FP-tree对频繁模式从下向上的挖掘,加快了挖掘过程[7]。随后,学者们又提出了一系列基于数据投影的算法,包括Han和Pei于2000年提出的FreeSpan算法[8]和2001年提出的PrefixSpan算法[9],这些都是经典的序列挖掘算法。近年来,在经典序列挖掘算法的基础上,学者们相继提出了优化的序列挖掘算法。2007年,张坤等提出一种基于GSP的MFS(Mining Frequent Sequence)算法[10],它不需要多次遍历数据库,通过连接不同长度的已知频繁序列来产生的候选序列,比GSP算法更高效;张利军等提出一种基于位置信息的序列模式挖掘算法PVS,在使用PrefixSpan算法时通过记录每个已产生的投影数据库的位置信息降低投影数据库的冗余,提高算法效率[11];2012年,刘栋等提出了基于Map Reduce的序列挖掘模式,在PrefixSpan算法的基础上,对模式挖掘任务进行分割,利用Map函数处理由不同前缀得到的序列模式并构造投影数据库,从而提高挖掘效率及简化搜索空间[12];2013年,吴信东等设计了一种带有通配符的模式挖掘算法One-Off Mining,通配符的加入使模式的形式更加灵活[13];2015年,刘端阳等首次在频繁序列模式挖掘算法中引入逻辑的思想,提出一种基于逻辑的频繁序列挖掘算法LFSPM,通过逻辑规则过滤,优化结果集[14]。
DNA序列挖掘作为序列挖掘领域最重要的应用之一,也获得了很多学者的特别关注。学者们结合DNA序列的特点后对传统的序列挖掘算法进行了有意识的改造提出了在处理DNA序列是更高效的算法。2007年,朱扬勇等对于DNA序列挖掘的问题进行了综述,回顾了DNA序列挖掘的发展历程,总结出DNA序列挖掘经历了三个阶段,依次是基于统计技术的应用阶段,一般化挖掘方法的应用阶段到如今的专门面向DNA序列挖掘的设计阶段[15];2007年,熊赟等提出了一种融合自底向上和自顶向下策略挖掘DNA重复序列的新算法DnaReSM[16],克服了完全自底向上算法产生大量短候选模式的缺点;2011年,周溜溜等提出了一种基于频繁子树挖掘策略的DNA频繁模式挖掘算法,绕开了传统序列的对比方式,将序列按照后缀树结构方式进行组织约减提升识别效率[17];2013年,姜华等在引入近似度的概念的基础上,构造频繁近似模式,提出了频繁近似模式的挖掘算法SFAP[18];2015年,毛国君等在分析DNA序列的特点的基础上,提出了一种基于关联矩阵(AMSMA)的算法,将扫描数据库得到的DNA序列信息储存在关联矩阵(Association Marix)中,利用矩阵压缩空间使用率,体现出较好的时间和空间效率[19]。这些算法关注于DNA序列的特点,大大提升了算法的效率。
本文中汲取了上面多个算法的精华,分别是PrefixSpan算法中基于前缀的思想[9]、PVS算法中位置信息的概念[11]和AMSMA算法中关联矩阵的概念[19],利用这些算法的优势提出一种高效的基于位置信息的DNA序列挖掘算法PBSMA(Position-Based Sequence Mining Algorithm)。
2 PBSMA算法描述
2.1 基本概念
定义1 DNA序列(DNA Sequence):给定字符集合E={A,T,C,G},一个DNA序列被表示为S=
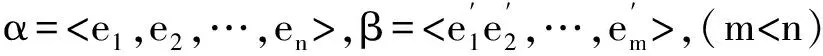
例如,在序列abacd中,aba是abacd的前缀,而ba不是。
定义3 关联矩阵:给定DNA序列S=
例如在序列s=
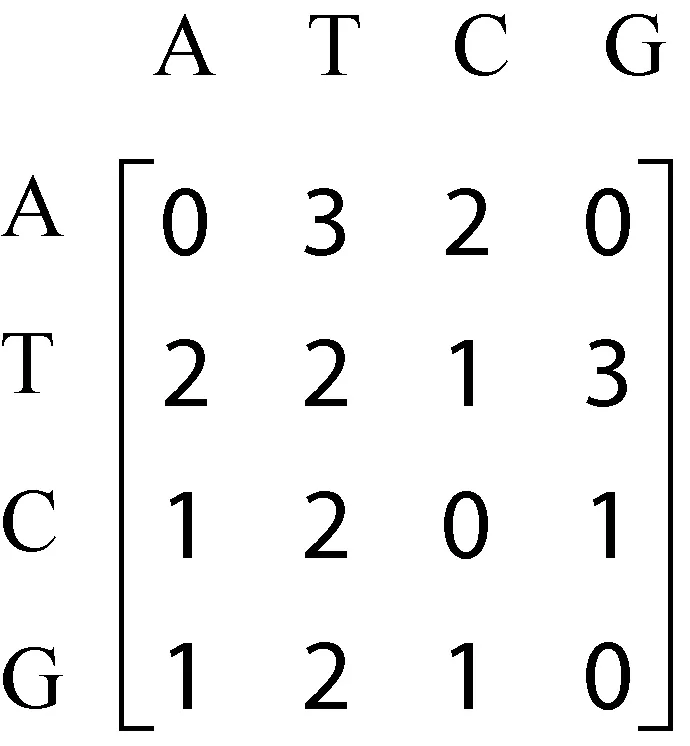
图1 关联矩阵举例
定义4 位置信息:一个子序列在数据库S中的位置信息为这个子序列在数据库S中的序列号和在序列中的结束位置,记为(s,v),其中s表示S中包含该模式序列的序号;v表示对应的序列关于该模式的结束位置。当一个子序列在数据库中多次出现时,这个子序列的位置信息表示为((s1,v1),(s2,v2),…,(sn,vn))。
2.2 算法思路
PBSMA算法是一种基于前缀的深度优先的挖掘算法,以DNA数据为例,在使用PBSMA算法处理上,首先通过扫描数据库来获取大1-项集L1={A,T,C,G},然后依次以L1中的元素为前缀构造关联矩阵,例如先以A为前缀产生所有以A开头的关键模式,深度优先直到不再有关键模式产生时,再以T为前缀……依次类推,直到遍历过L1中的所有元素后停止。
PBSMA算法的步骤如下:
(1) 扫描序列数据库,得到长度为1的序列模式L1,作为初始的种子集;
(2) 对于L1中的每一个元素,依次取出一个元素作为新候选模式的前缀,以此来划分搜索空间;
(3) 将上一步得到的前缀作为关联矩阵的行,以L1中的所有元素作为关联矩阵的列,生成关联矩阵。计算关联矩阵中每一个候选模式在数据库中出现的次数(即支持度)并保存每次出现的位置信息;
(4) 检测关联矩阵中候选模式的支持度是否大于设定的最小支持度,若大于,则该子序列为关键序列,加入到L2并更新其位置信息;若不大于,则不是关键序列,删除之前保存的位置信息;
(5) 利用L2和位置信息依次生成更高阶关联矩阵,得到L3,L4……直到没有新的序列模式或新的候选序列模式产生为止。
重复步骤(2)-步骤(5),直到遍历完L1中的所有序列。
值得注意的是,与从单条序列中挖掘关键模式不同,在多条序列的挖掘中,如果某子序列在一条记录中多次出现,仍然只计数一次,但需要记录该子序列出现的每一个位置信息,后面的例子中将会详细说明这一点。
2.3 算法举例
例1 例如在表1中的DNA序列数据库,设最小支持度为2,则子序列AT是一个频繁模式,它的位置记为((1,2)、(1,7)、(2,4)、(2,9)、(3,4))。其中在(1,2)中,1表示S中的第一条序列,2中表示AT这个子序列的最后一个字符在序列1中所处的位置为2,后四者亦同。

表1 例1中DNA序列数据库
在上例中,首先构造A为前缀的关联矩阵,构造关联矩阵如图2所示。
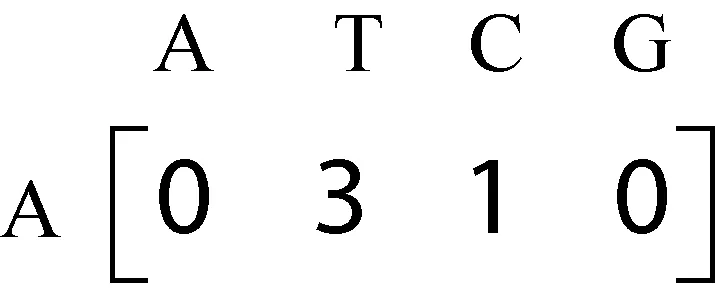
图2 前缀为A的一阶关联矩阵
扫描计数的同时,记录到的AT的位置信息(1,2)、(1,7)、(2,4)、(2,8)和(3,4),AC的位置信息(3,7)。当支持度为2时,AT是频繁模式,而AC不是,因此进行剪枝,不再保留AC的位置信息。
利用上一步得到的长度为2的频繁模式构造2阶关联矩阵。此时无需扫描数据库,利用上一步得到的位置信息,直接去序列中寻找AT的后缀,构造关联矩阵并计数,同时更新位置信息。在上例中,出现在(1,2)位置的AT直接后缀为T,则在关联矩阵相应位置将子序列ATT的支持度增加1,并保存位置信息(1,3),出现在(2,4)位置的AT直接后缀为C,则子序列ATC的支持度增加1,并保存位置信息(2,5)…依次找到每一个位置信息下的AT的后缀形成关联矩阵并保存位置信息,得到如图3的二阶关联矩阵。
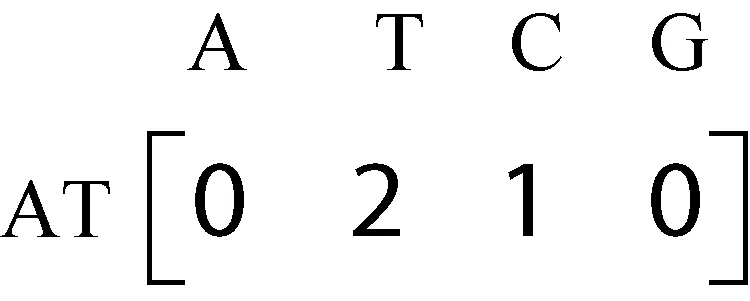
图3 以A为前缀的二阶关联矩阵
对应位置信息为ATT(1,3)、(1,8)、(3,5),ATC(2,5)。在支持度为2时,得到长度为3的频繁序列ATT,删除ATC的位置信息。同样,利用长度为3的频繁序列构造4阶关联矩阵,利用位置信息找到长度为4的候选序列,计数并记录位置信息后发现长度为4的频繁模式ATTA,位置信息为(1,9)、(2,6),如图4所示。利用长度为4的频繁模式构造五阶关联矩阵,如图5所示,没有发现支持度大于2的频繁模式。至此,以A为前缀的频繁模式全部找到。
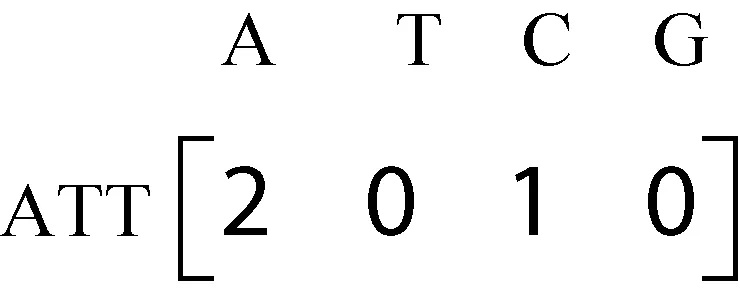
图4 以A为前缀的三阶关联矩阵
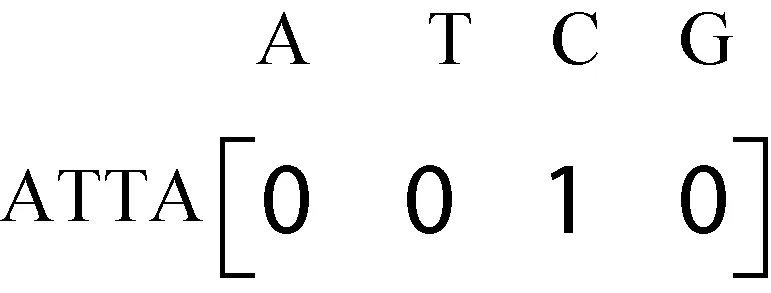
图5 以A为前缀的四阶关联矩阵
同样的,应用上面的方法分别建立以T、C、G为前缀的各阶关联矩阵,采用深度优先的原则分别发现以T、C、G为前缀的频繁模式。产生的所有频繁模式如表2所示。

表2 例1中的所有频繁模式
由此可得到最大关键模式有:CTA,TCG,CGATTA。
2.4 算法伪代码
算法描述:PBSMA算法伪码
输入:稀疏项目集序列数据库S;最小支持度min-sup
输出:S的关键子序列KS
1. scan S to get L1{};
//扫描数据库找到所有的频繁1-序列
2. FOR m=0 TO L1.size()
//深度优先,最外层循环依次遍历L1中的每个序列
3. former=L1.get(m);
4. FOR n=0 TO L1.size();
//该层循环意在获取初始位置
5. later=L1.get(n);
6. string=former+later;
//子序列连接
7. Calculate support of string occur in S ;
//计算子序列支持度
8. IF(support >=min_sup)
//大于等于最小支持度为频繁模式
9. add string to KS;
10. save Position of string;
//记录子序列位置信息
11. WHEN(Position is not empty) DO
12. use Position to extend longer subsequence;
13. calculate support;
//利用位置信息寻找更长的频繁模式并计算支持度
14. IF support>=min-sup
15. add subsequence to KS;
16. update Positon;
//更新位置信息
17. ELSE
18. delete Position;
//不满足最小支持度的序列删除位置信息
19. END DO
20. Retrun KS
//输出所有频繁序列
3 实验分析
本部分实验中DNA数据来源于美国国家生物技术信息中心(http://www.ncbi.nim.nih.gov)。由于这里只探讨算法的可用性和时间空间效率,而不是要解决生物问题,也无需对实验结果进行解释,因此为了更好地控制实验变量,实验数据在原始数据上进行了一定的处理,具体处理方法在下面的各个实验中作了说明。
实验是在一台4GB内存、使用英特尔酷睿i3,1.40 GHz处理器的计算机上进行。采用的对比算法是同样基于分治思想的PrefixSpan算法[9]。实验的目标主要是检测本文算法的精度和效率(包括时间效率和空间效率)。
实验1 比较内存空间使用情况
由于PBSMA算法不需要产生投影数据库,因此占用更少的内存。实验1模拟PreifixSpan算法和PBSMA算法的运行过程,对实验1中的序列数据进行分析,每一步的占用内存如表3所示。由于两个算法都是基于前缀的分治思想,因此表3中先讨论以L1中频繁1-序列为前缀的情况,最后再从总体上进行讨论。
值得注意的是,PrefixSpan算法的投影数据库保存投影序列,在本实验中即字符序列,是字符型数据;PBSMA算法的位置信息保存坐标位置,由两个整型数值构成,为方便表示,在表3的讨论中均将其作为字符处理。

表3 PrefixSpan算法和PBSMA算法的内存使用情况
从表3的实验结果可以看出,无论利用分治的思想在每一个小范围内讨论,还是从总体上考虑最大使用内存和合计使用内存情况,PBSMA算法的空间效率都要好于PrefixSpan算法。虽然PBSMA算法需要记录较多的位置信息,但由于每个位置信息仅占用两字符的内存,相比于PrefixSpan算法投影数据库中的子序列相比,仍然有优势。
实验1中序列数据量小,并且每条序列长度较短,当应用于实际的序列数据库时,PrefixSpan算法将产生更多的投影数据库,因而PBSMA算法的空间优势将会更明显。
实验2 比较不同的最小支持度下执行时间
本实验的数据来源于美国国家生物技术信息中心(NCBI)的序列数据库,序列被随机划分为长度20~30的序列,实验记录1 369条,共28 739个字符,将此实验数据集命名为PBSMA实验数据集-1。
利用PBSMA实验数据集-1,在两个最小支持度区间进行实验,分别是小支持度区间(5%~30%)、大支持度区间(10%~90%),测试本文提出的PBSMA算法和PrefixSpan算法的运行效率。表4给出的是实验数据在小支持度区间范围内挖掘出的频繁序列的个数,图6对应给出在该区间内PrefixSpan和PBSMA运行时间效率的对比;表5给出的是实验数据在大支持度区间范围内挖掘出的频繁序列的个数,图7对应给出在大支持度区间PrefixSpan和PBSMA运行时间效率的对比。
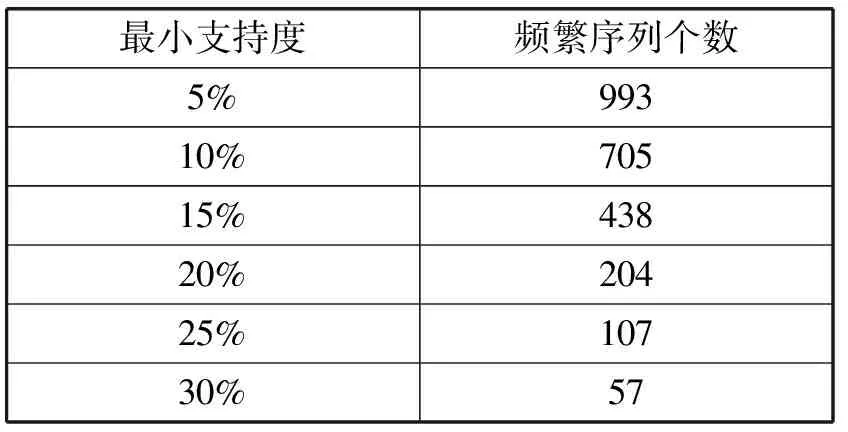
表4 小支持度区间内挖掘出频繁序列个数

图6 小支持度区间内算法时间效率对比
由表4和图6可以看出,在小支持度区间,实验数据集-1产生较多的关键模式。随着最小支持度的增加,PBSMA和PrefixSpan算法的执行时间都在下降,这是因为随最小支持度的增加,生成的关键子序列在减少,并且在该数据集中,关键子序列减少的速度很快。同时也可以看出,PBSMA算法在处理DNA序列时时间效率明显高于PrefixSpan算法。
表5和图7从更宏观、更全面的角度进行实验,展现了最小支持度在更大范围内变化时两算法的执行效率对比。实验结果表明:随着最小支持度的增加,PBSMA和PrefixSpan算法的执行时间都在下降,同时,PBSMA算法在各支持度下的时间效率明显都要高于PrefixSpan算法。其中,在支持度在10%~30%的范围内变化时,由于挖掘出的频繁子序列数量减少速度很快,因此算法执行时间下降速度很快;在40%~70%的范围内,频繁子序列数量减少速度相对较慢,因此挖掘算法的执行时间降速放缓甚至趋于平稳;然而在70%及以上的支持度下,由于此时已经不再有频繁模式被挖掘出来,算法执行时间再一次骤减,并且由于两算法在扫描一次数据库后基本不用再进行后面的循环,因此两算法的运行时间都趋于0,PBSMA算法的优势也不如产生大量频繁子序列时明显。
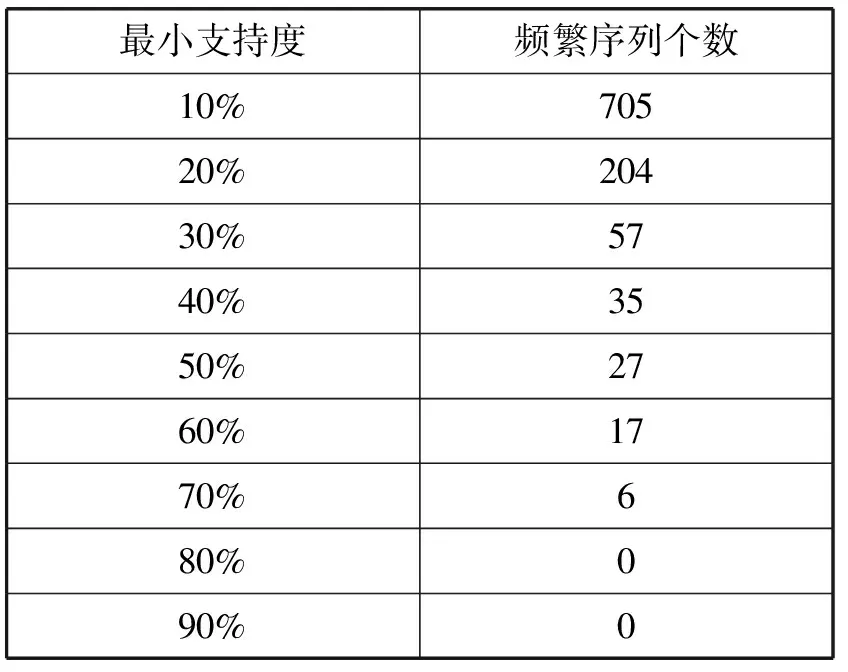
表5 大支持度区间内挖掘出频繁序列个数
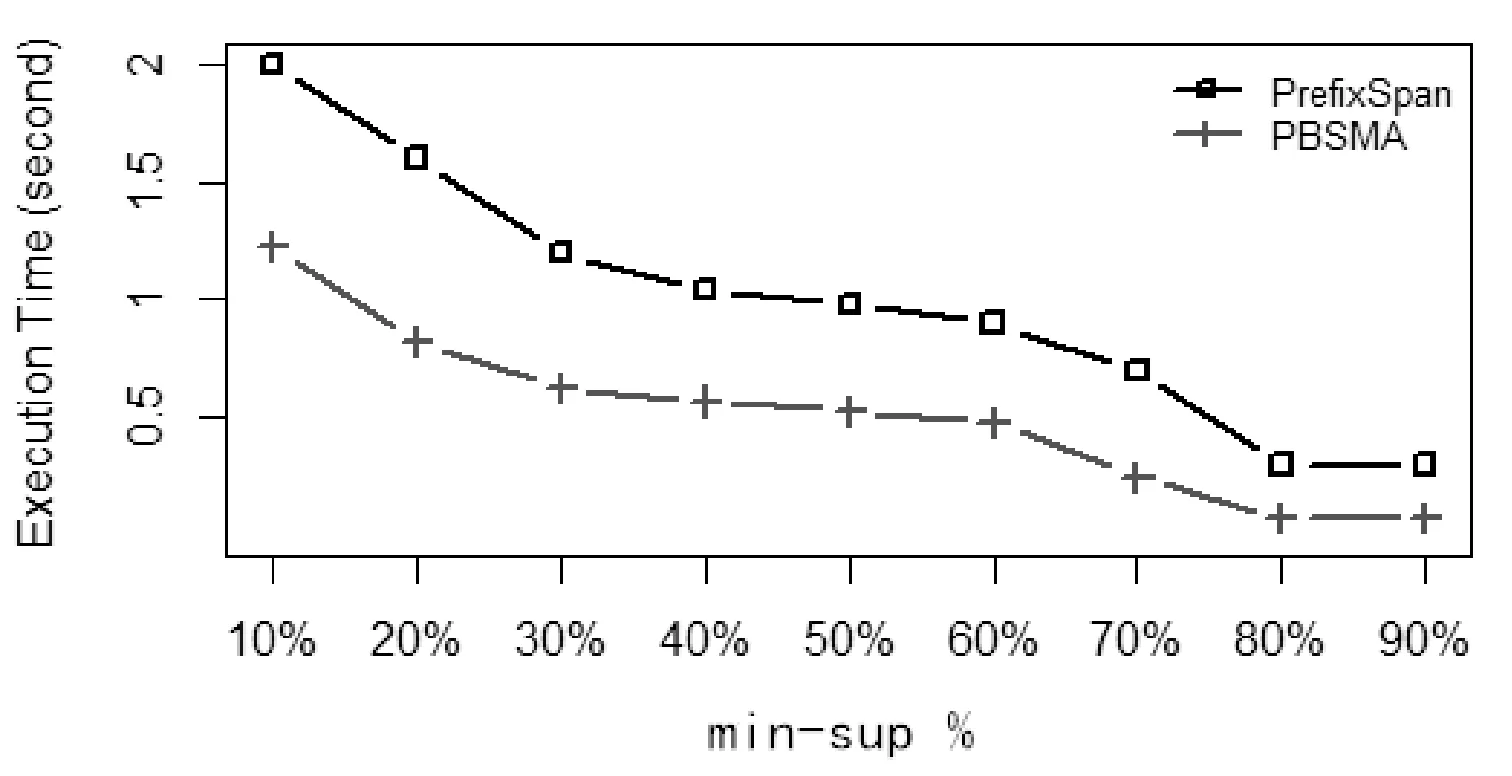
图7 大支持度区间内算法时间效率对比
实验3 比较不同的DNA序列数下执行时间
本实验的数据来源于美国国家生物技术信息中心(NCBI)的序列数据库,创建5个数据文件,其中存放序列数量分别为2 000、4 000、6 000、8 000、10 000,每条序列长度20~30。将此实验数据集命名为PBSMA实验数据集-2。
本实验在控制最小支持度变量为20%不变的前提下,运用PBSMA实验数据集-2中五个不等长数据文件,考察随着序列数量攀升的情况下两算法的执行效率。实验结果如图8所示。
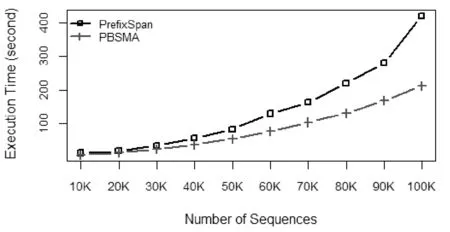
图8 序列数量增加时执行时间的比较
图8表明:在固定的最小支持度下,随着序列数据库规模的增大,即序列数量的增加,PBSMA和PrefixSpan算法的执行时间在攀升。在数据量较小的情况下,两算法的运行时间效率相差不多,但在数据量逐渐增多的过程中,PBSMA展现出越来越优于PBSMA算法的表现,特别地,当序列数量为100 K时,PrefixSpan算法所需要的时间几乎是PBSMA算法运行时间的两倍,并且可以预见的是,当数据量更大时,PBSMA算法的优势将更加明显。因此可以得出结论,在任何数据量下,PBSMA的算法运行效率都要优于PrefixSpan算法,并且数据量越大,PBSMA算法在时间效率上的提升越明显。
4 结 语
本文提出了基于位置信息的序列挖掘算法——PBSMA算法。PBSMA算法结合了关联矩阵、基于前缀、位置信息等的思想,用于从多条序列中挖掘出频繁模式。较之PrefixSpan算法,PBSMA算法有如下的优点:
(1) 不需要产生投影数据库,大大节约了存储空间;
(2) 由位置信息直接定位到序列数据库中,不需要多次扫描数据库及投影数据库,减少时间花销。
在实验中,PBSAM算法展现出高于PrefixSpan算法的时间和空间效率,证明了算法的有效性和高效性。但是,本文提出的序列挖掘算法在时间和空间上都还有提升的空间,算法也可以寻找更广阔的应用空间,这都是未来应该着重研究的领域。
[1] Agrawal R,Srikant R.Mining sequential patterns[C]//Proceedings of the 11th International Conference on Data Engineering.IEEE Computer Society,1995:3-14.
[2] 毛国君,段丽娟,王实,等.数据挖掘原理与算法[M].2版.北京:清华大学出版社,2007:217-218.
[3] 陈卓,杨炳儒,宋威,等.序列模式挖掘综述[J].计算机应用研究,2008,25(7):1960-1963,1976.
[4] 罗春雨,毛国君,邱洪君.序列分析技术在DNA序列挖掘中的应用[J].计算机系统应用,2005(12):22-25.
[5] Agrawal R,Imieliński T,Swami A.Mining association rules between sets of items in large databases[N].ACM SIGMOD Record,1993,22(2):207-216.
[6] Srikant R,Agrawal R.Mining sequential patterns:generalizations and performance improvements[C]//Proceedings of the 5th International Conference on Extending Database Technology:Advances in Database Technology,1996:3-17.
[7]HanJ,PeiJ,YinY.Miningfrequentpatternswithoutcandidategeneration[C]//Proceedingsofthe2000ACMSIGMODInternationalConferenceonManagementofData.NewYork,NY,USA:ACMPress,2000:1-12.
[8]HanJ,PeiJ,Mortazavi-AslB,etal.FreeSpan:frequentpattern-projectedsequentialpatternmining[C]//Proceedingsofthe6thACMSIGKDDInternationalConferenceonKnowledgeDiscoveryandDataMining.NewYork,NY,USA:ACMPress,2000:355-359.
[9]PeiJ,HanJ,Mortazavi-AslB,etal.PrefixSpan:miningsequentialpatternsefficientlybyprefix-projectedpatterngrowth[C]//Proceedingsofthe17thInternationalConferenceonDataEngineering.IEEEComputerSociety,2001:215-224.
[10] 张坤,朱扬勇.无重复投影数据库扫描的序列模式挖掘算法[J].计算机研究与发展,2007,44(1):126-132.
[11] 张利军,李战怀,王淼.基于位置信息的序列模式挖掘算法[J].计算机应用研究,2009,26(2):529-531.
[12] 刘栋,尉永清,薛文娟.基于MapReduce的序列模式挖掘算法[J].计算机工程,2012,38(15):43-45.
[13] 吴信东,谢飞,黄咏明,等.带通配符和One-Off条件的序列模式挖掘[J].软件学报,2013,24(8):1804-1815.
[14] 刘端阳,冯建,李晓粉.一种基于逻辑的频繁序列模式挖掘算法[J].计算机科学,2015,42(5):260-264.
[15] 朱扬勇,熊赟.DNA序列数据挖掘技术[J].软件学报,2007,18(11):2766-2781.
[16] 熊赟,陈越,朱扬勇.DnaReSM:一个基于多支持度的DNA重复序列挖掘算法[J].计算机科学,2007,34(2):211-212,封四.
[17] 周溜溜,业宁,徐昇,等.基于频繁子树挖掘的DNA重复序列识别方法[J].微电子学与计算机,2011,28(9):193-196,201.
[18] 姜华,孟志青,周克江.DNA序列频繁近似模式挖掘[J].生物信息学,2013,11(1):11-15.
[19] 毛国君,杨静欣.一种基于关联矩阵的高效DNA序列挖掘算法[J].计算机科学与应用,2015,5(8):271-277.
AN EFFICIENT POSITION-BASED DNA SEQUENCE MINING ALGORITHM
Yang Jingxin Mao Guojun
(SchoolofInformation,CentralUniversityofFinanceandEconomics,Beijing100081,China)
Similar to Apriori algorithm in generating frequent patterns need to scan the database several times, and generate a large number of candidate sets. Algorithms based on the projection database, such as FreeSpan and PrefixSpan, in generating frequent patterns will produce a large number of projection database, taking up a lot of memory space, which have caused a lot of redundancy. Aiming at the shortcomings of the previous sequence mining algorithms, an efficient sequence mining algorithm named PBSMA is proposed in this paper. The PBSMA reduces the scanning of the database by recording the position information of frequent subsequences, and gradually enlarges the length of the frequent patterns by using the position information. The algorithm uses the idea of association matrix and the concept of prefix in PrefixSpan algorithm to search for a longer key pattern. The experimental results show that the PBSMA is more efficient than PrefixSpan algorithm both in time and space.
Sequence mining DNA sequence Position information Association matrix Prefix
2016-05-17。国家自然科学基金项目(61273293)。杨静欣,硕士生,主研领域:数据挖掘和生物信息学。毛国君,教授。
TP311.1
A
10.3969/j.issn.1000-386x.2017.06.042
