动态-静态混合的时序蛋白质网络构建方法
2016-12-22代启国郭茂祖刘晓燕王春宇
代启国, 郭茂祖, 刘晓燕, 王春宇
(1.哈尔滨工业大学 计算机科学与技术学院, 哈尔滨 150001; 2.大连民族大学 计算机科学与工程学院, 辽宁 大连 116600;3.大连市民族文化数字化重点实验室(大连民族大学), 辽宁 大连116600)
动态-静态混合的时序蛋白质网络构建方法
代启国1,2,3, 郭茂祖1, 刘晓燕1, 王春宇1
(1.哈尔滨工业大学 计算机科学与技术学院, 哈尔滨 150001; 2.大连民族大学 计算机科学与工程学院, 辽宁 大连 116600;3.大连市民族文化数字化重点实验室(大连民族大学), 辽宁 大连116600)
目前已公开的蛋白质网络多为静态网络,不能有效描述细胞中蛋白质的动态活动特点. 通过融合基因表达数据,研究人员可以构建出描述蛋白质动态性的时序蛋白质网络. 现有方法假设所有蛋白质都是动态变化的,而事实上除动态蛋白质外细胞中还包含相对稳定的静态蛋白质. 为此,提出了一种基于动态-静态蛋白质混合的时序网络构建新方法. 该方法根据基因表达变化情况将蛋白质分为动态和静态两类,并在构建各时刻网络时考虑动态与静态蛋白质之间的相互作用关系. 实验结果表明,利用本文方法构建的时序蛋白质网络可以提高蛋白质复合体识别的准确性,从而验证了本文方法的可行性.
时序蛋白质网络;蛋白质相互作用;基因表达;生物网络;蛋白质复合体识别
蛋白质相互作用网络(protein-protein interaction network,简称蛋白质网络)是描述细胞中蛋白质活动的一种生物网络[1-2]. 由于蛋白质在生命活动中具有十分重要的作用,因而基于蛋白质网络的分析和挖掘方法越来越受到关注. 蛋白质复合体识别[3-5]、关键蛋白质识别[6]等很多生物信息研究都是以蛋白质网络为基础的.
现有公开蛋白质网络通常可表示为一个图,其中节点表示蛋白质,边表示蛋白质之间的相互作用关系. 这种网络一般假设蛋白质的丰度都是静态不变的,所描述的蛋白质之间相互作用也是持续稳定的,因而称为“静态蛋白质网络”. 然而,真实细胞系统中蛋白质及其相互作用是随时变化的,具有一定的动态性. Lichtenberg等人研究发现,大部分复合体都是由动态和静态两类蛋白质构成[7]. 然而,静态蛋白质网络的简单图结构无法描述蛋白质动态特性[8-10],因此有必要构建能够描述蛋白质动态特性的时序蛋白质网络模型.
在构建时序蛋白质网络的过程中,一个关键问题是如何确定蛋白质在何时处于激活状态. 采用单一固定阈值方法确定蛋白质的活动状态与非活动状态虽然简单[8],但存在一定局限. 例如,一些蛋白质的含量在整个细胞周期都较低. 如果采用单一的固定阈值,那么这些蛋白质可能会被认为是非激活状态而被忽略掉. Hegde等人提出了利用不同基因表达数据中表达值的均值确定蛋白质的活动状态[10]. 王建新等人[11]针对蛋白质不同的表达模式特性,提出了3-σ阈值计算方法. 上述方法假设所有蛋白质丰度都是动态变化. 然而,生物学研究表明,细胞中蛋白质并非全部都是动态的,有些蛋白质的丰度受细胞周期、环境等影响较小,表现出一定的静态特性[12]. 因此,将所有蛋白质均视为动态的方法,不能准确地刻画细胞中真实的蛋白质活动规律.
为此,本文提出一种动态-静态蛋白质混合的时序蛋白质网络构建方法(A hybrid temporal protein network,HTPIN). 该方法首先将蛋白质划分为动态和静态两类,然后同时利用动态蛋白质与静态蛋白质之间相互作用关系构建时序网络. 通过相关实验测试与分析,利用本文方法所构建的时序蛋白质网络具有很好的可靠性,可以提高蛋白质复合体识别的准确性.
1 动态-静态混合的时序网络构建方法
给出一种基于动态-静态蛋白质混合的时序蛋白质网络构建方法(HTPIN). 首先,以基因表达数据为基础,利用蛋白质编码基因在不同时刻表达值变化的波动性,将蛋白质划分为动态和静态两类;然后,采用3-σ阈值[11]判断动态蛋白质激活时刻;最后,构建各时刻蛋白质网络. 该方法的基本流程如图1所示.
1.1 动态与静态蛋白质
以基因表达量作为判定蛋白质动、静态属性的依据. 假设给定蛋白质网络中有N个蛋白质,基因表达数据中记录了T个时间点的表达量. 对于蛋白质i(1≤i≤N),其对应编码基因在所有时刻的表达值Expi可表示为
其中,eit表示蛋白质i的编码基因在t时刻的表达量. 则蛋白质i表达量的方差[12]为
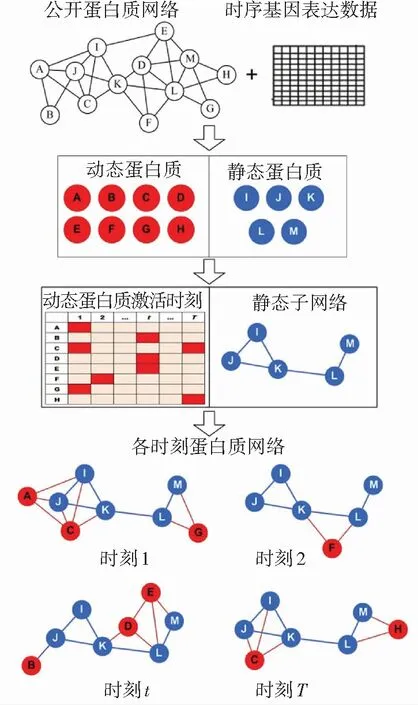
图1 HTPIN方法构建时序蛋白质网络过程示意
Fig.1 Illustration of construction process of temporal protein networks with HTPIN
利用表达量波动值EVi,表示蛋白质i在所有时刻表达量的方差值. 具体来讲,表达量方差值高于蛋白质i表达量方差的蛋白质数目记为ni,则蛋白质i编码基因表达量波动值EVi=ni/N. 该值介于0和1之间,越接近于0表明蛋白质表达量越稳定;越接近于1则表示蛋白质表达量波动性越大.
引入波动性阈值thDyn(0≤thDyn≤1)确定每个蛋白质的动、静态属性. 蛋白质i的波动值EVi大于或等于阈值thDyn,则认为该蛋白质是动态蛋白质,否则为静态蛋白质,具体可形式化为
其中isDyn(i) 值为1表示蛋白质i是动态蛋白质,为0表示蛋白质是静态蛋白质. 动态阈值thDyn为HTPIN算法参数.
1.2 确定蛋白质激活时刻
1.2.1 动态蛋白质的激活状态
动态变化的蛋白质只有在其处于激活状态时,才与其他蛋白质发生相互作用. 因此,需要确定动态蛋白质处于激活状态的时间点. 动态蛋白质在不同时间点对应表达量具有很大的波动性. 对于某一时刻,如果一个动态蛋白质的表达量相对较高,则认为其处于活动状态;而表达量相对较低,则认为是非活动状态. 为此,引入激活阈值thiAct来判断动态蛋白质在各时间点所处的状态. 对于动态蛋白质i(isDyn(i)=1),如果其在时间点t对应基因的表达值eit高于该激活阈值thiAct,则认为其在t时刻为激活状态,否则为未激活状态,即
其中,isActive(i, t)值为1表示激活,0表示未激活. 采用3-σ方法[11]确定激活阈值thiAct,其具体方法为
1.2.2 静态蛋白质的激活状态
静态蛋白质对应的基因表达量相对稳定,因而通常认为它们在整个生物过程中活动状态是持续稳定的[7, 12]. 因此,静态蛋白质在所有时刻均处于激活状态.
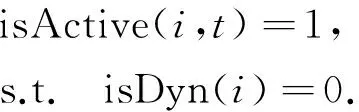
1.3 构建时序蛋白质网络
基于动态-静态蛋白质混合的时序网络构建方法主要分为两个步骤:首先,抽取静态蛋白质之间相互作用网络,即静态子网络;然后,以其为基础在每个时间点增加在该时刻处于激活状态的动态蛋白质及其相互作用关系.
1.3.1 输入数据描述
输入数据包括公开蛋白质网络G和某个生物过程中的时序基因表达数据Exp. 其中,网络G可描述为G=(V, E),V表示其中蛋白质集合,E表示蛋白质相互作用关系集合. 网络中的蛋白质数目记为N(N=|V|). 时序基因表达数据Exp中收录了所有N个蛋白质在T个时刻下对应基因的表达量,即

1.3.2 抽取静态子网络
抽取静态子网络是指获取输入蛋白质网络G中所有静态蛋白质构成的子网络. 通过预先抽取静态子网络可以避免在各时刻重复构建,提高构建时序网络的效率. 所抽取的静态子网络可表示为GStat=(VStat, EStat),其中包含所有静态蛋白质VStat和它们在G中的相互作用关系EStat.
1.3.3 构建各时刻动态-静态混合网络
时序蛋白质网络GTC可表示为
(8)
其中,Gt为t时刻蛋白质网络,表示为Gt=(Vt,Et),Vt和Et分别构成该时刻网络的蛋白质和相互作用关系集合. 由于已经预提取了每个时刻静态子网络,因此构建各时刻网络可以转化为在静态子网络基础上,增加在该时刻处于激活状态的动态蛋白质以及与它们之间的相互作用关系;此外,还增加它们与静态蛋白质之间的作用关系.
1.3.4 动态-静态混合时序蛋白质网络构建算法
以上述主要过程为基础,基于基因表达数据的动态-静态混合时序蛋白质网络的构建算法(HTPIN)的描述如下.
输入:蛋白质相互作用网络G=(V,E);基因表达数据Exp={Exp1,…,Exp|V|};动态阈值thDyn.
输出:混合型时序蛋白质网络GTC.
初始化静态子网络GStat=(VStat,EStat),VStat=∅,EStat=∅;
初始化动态蛋白质集合VDyn=∅;
for each p in V/* 将蛋白质分为动态和静态两类蛋白质 */
if p为动态蛋白质isDyn(p,thDyn)
将p加入到动态蛋白质集合VDyn=VDyn∪{p};
else
将p加入到静态蛋白质集合VStat=VStat∪{p};
end if
end for
for each epqin E
if both p and q in Estat
将epq加入到EStat;
end if
end for
初始化时序网络GTC={G1,…,GT}
for t=1 to T/* 获取各时刻蛋白质网络 */
初始化t时刻网络Gt=(Vt,Et),Vt=∅,Et=∅;
for each p in VDyn/* 获取t时处于激活状态的动态蛋白质 */
if 蛋白质p在t时刻处于激活状态isActive(p,t)
Vt=Vt∪{p};
end if
end for
for each p in Vt/* 获取激活动态蛋白质之间的相互作用 */
for each q≠p in Vt
if 输入网络G中存在p和q的相互作用,即epqin E
将epq加入到Et;
end if
end for
end for
for each p in Vt/* 激活动态蛋白质与静态蛋白质相互作用*/
for each q in VStat
if 输入网络G中存在p和q的相互作用,即epqin E
将epq加入到Et;
end if
end for
end for
Vt=Vt∪VStat;
end for
输出时序蛋白质网络GTC
2 实验结果
为了验证本文HTPIN方法的合理性,对其进行多方面的测试. 基于蛋白质网络识别复合体是针对蛋白质网络进行信息挖掘的典型问题. 蛋白质网络是计算识别复合体的重要数据依据. 蛋白质复合体是由多个蛋白质通过物理相互作用结合形成,这些相互作用紧密的蛋白质一般在蛋白质网络中对应稠密的子图结构[1-2,13]. 因而,可以通过挖掘网络中稠密子图识别蛋白质复合体. 也就是说,从蛋白质网络中识别复合体是一个图聚类问题. 因此,本文通过分析所构建的时序蛋白质网络是否可以提高复合体识别准确性来评价HTPIN方法的有效性.
2.1 实验设计及实验数据
为了达到通过识别复合体验证时序网络的目的,在实验中采用了如下方案:对于给定的一种复合体识别算法和构建的时序网络GTC={G1,G2,…,Gt,…,GT},首先将识别算法依次在每个时刻的蛋白质网络Gt上运行,获得在t时刻的识别结果Ct;然后将在所有时刻网络上的识别结果集合在一起,去除重复复合体,形成复合体集合C,即为该识别算法在时序网络GTC上最终识别结果. 通过分析识别结果C与已知复合体集合(CYC2008)[14]匹配的组合评分,以及功能一致性两方面评价时序网络. 组合评分具体为F-Measure、ACC和MMR三项子指标之和[15]. 为了更加客观地进行评价,避免个别算法对时序网络的偏好性,在实验中测试了CPL[16]、ClusterONE[17]和PLSMC[15]3种不同类型算法.
在基因表达数据方面,采用了GEO数据库中与酵母代谢周期相关的GSE3431系列数据[18]. 该数据采集背景是限制营养条件下连续培养酵母细胞的3个代谢周期,每个周期采样12个时间点,间隔为25 min.
在静态蛋白质网络方面,采用了酵母的BioGRID蛋白质网络[19]. 在两种类型数据之间,采用ORF命名类型来作为关键字以建立两种数据中的基因与蛋白质之间的对应关系. 滤掉两种数据中未建立映射关系的基因或蛋白质.
2.2 波动性阈值的影响
HTPIN时序网络构建方法的核心是动态-静态蛋白质混合的思想. 为了验证这种方案的合理性,本文研究了控制静态与动态蛋白质混合比例的波动性阈值thDyn对所构建网络的影响. 测试了该阈值thDyn={0.0,0.1,…,0.9,10}的不同取值情况. 对于每个取值,统计分析不同算法在时序网络上的复合体识别结果.
图2描述了CPL、ClusterONE和PLSMC三种算法在采用不同波动性阈值而构建的时序网络上识别复合体结果的对比情况. 当thDyn=0时表明所有蛋白质均被视为动态蛋白质,并认为这些蛋白质只在部分时刻处于活动状态;而当thDyn=1时则表示所有蛋白质都被视为静态蛋白质,在所有时刻均出现.
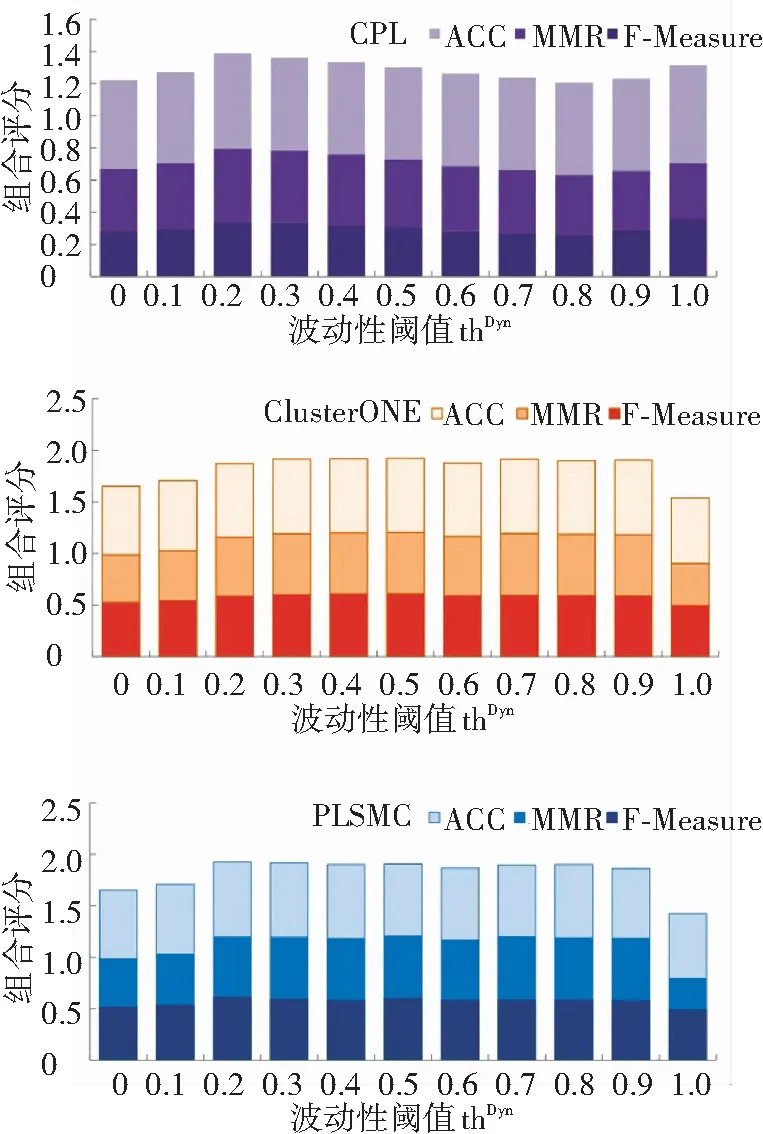
图2 采用不同波动性阈值thDyn构建时序网络对复合体识别性能的影响
Fig.2 The performance comparison of complex detection on time-course protein network constructed by HTPIN with different volatility threshold thDyn
从图2可见,不同算法识别结果的组合评分均随波动性阈值thDyn的改变而较大发生变化. 但所有参与测试的算法识别结果的组合评分峰值均出现在当thDyn=0.2至0.4之间,即此时复合体识别结果要好于thDyn=0和thDyn=1两种极端情况. 由此表明,在构建时序蛋白质网络时,将动态蛋白质与静态蛋白质控制在合理的比例可以有效提高复合体识别的总体性能. 上述实验结果表明,本文提出的基于静态-动态蛋白质混合思想构建时序蛋白质网络的方法具有一定的合理性.
图3描述了在波动性阈值thdyn=0.2条件下,YKL101w、YNL286w和YLR011w3个静态蛋白质及其相邻接的部分动态蛋白质,在时刻1、4、7、10等4个时间点所构成的蛋白质网络. 从4个示例网络中可见,在每个时刻,与指定的静态蛋白质邻接的动态蛋白质有所差别,既体现了静态蛋白质的稳定性,同时也刻画了动态蛋白质的变化特点.
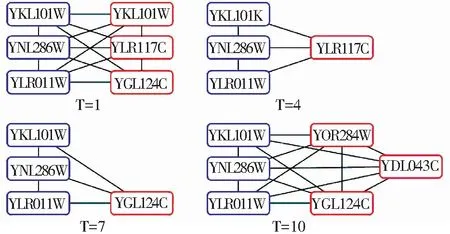
图3 在thDyn=0.2条件下,第1、4、7和10时刻点蛋白质子网络示意图
Fig.3 Sub-networks on time 1, 4, 7 and 10 under the condition of thDyn=0.2
2.3 不同类型蛋白质网络的对比
下面将HTPIN方法构建的时序网络、同类方法DPIN方法[11]所构建的时序网络进行比较. DPIN与HTPIN均是用来构建时序蛋白质网络的方法. 二者主要区别在于,DPIN方法将所有蛋白质均视为动态蛋白质,而HTPIN方法则根据表达量波动情况将蛋白质划分为动态、静态两类,并在此基础上构建混合蛋白质网络. 通过比较两类方法可以证明本文提出的基于“混合”思想构建时序网络方法的有效性. 此外,还与原始蛋白质网络进行了对比分析. 实验中,HTPIN方法采用了2.2节中获得最优组合评分的波动性阈值.
2.3.1 识别复合体与已知复合体匹配评分
表1列出了CPL、ClusterONE以及PLSMC三种算法在不同类型蛋白质网络上识别复合体的评价指标,其中组合评分为F-Measure、ACC和MMR三项之和[15]. 在HTPIN和DPIN构建的网络上,ClusterONE和PLSMC算法识别结果的组合评分要好于在原始蛋白质网络评分值,表明通过融合基因表达数据构建时序蛋白质网络可以提高蛋白质复合体识别的准确性的. 值得注意的是,所有参与测试的复合体识别算法在HTPIN方法构建的网络上均取得了最优的组合得分,表明本文方法较DPIN方法更有利于复合体的识别.
2.3.2 识别复合体的功能一致性分析
下面对复合体识别算法在不同类型网络上识别复合体的GO功能一致性进行了分析. 利用GOTermFinder工具[20]分析所识别复合体的功能一致性. 实验中计算了在p-value≤1.0E-10时功能一致性的复合体占所有识别复合体的比例. 该比例越高,表明所识别复合体功能一致性越高,具有更强的生物意义.
表1 不同蛋白质网络识别的复合体与已知复合体匹配
Tab.1 Comparison of matching between known complexes and the complexes detected by using different protein networks
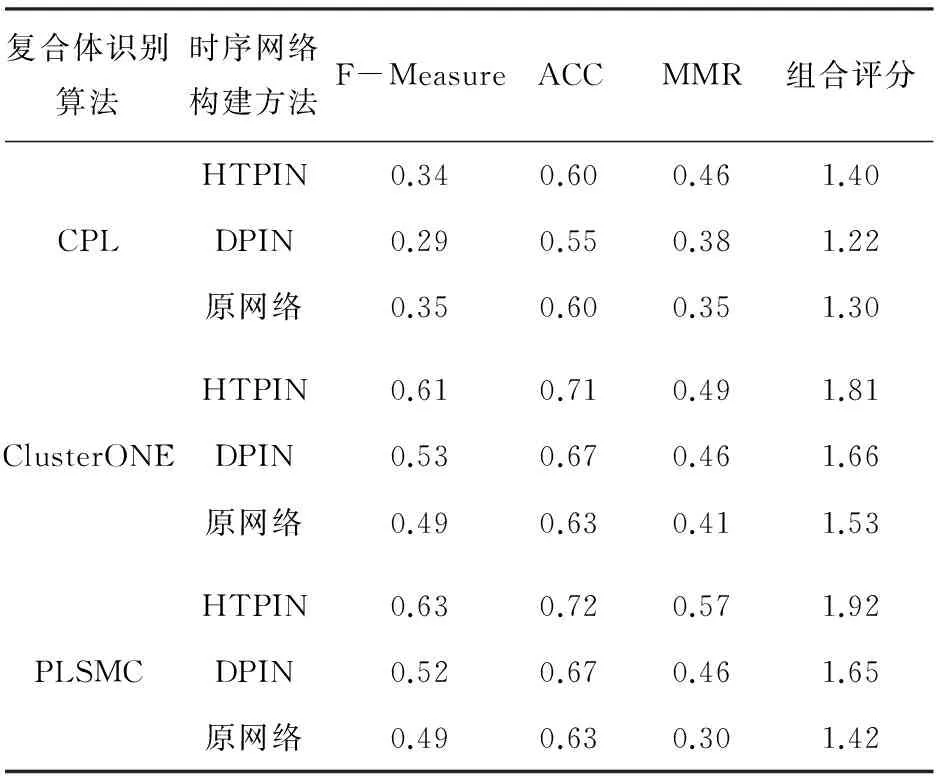
复合体识别算法时序网络构建方法F-MeasureACCMMR组合评分HTPIN0.340.600.461.40CPLDPIN0.290.550.381.22原网络0.350.600.351.30HTPIN0.610.710.491.81ClusterONEDPIN0.530.670.461.66原网络0.490.630.411.53HTPIN0.630.720.571.92PLSMCDPIN0.520.670.461.65原网络0.490.630.301.42
表2 不同蛋白质网络下识别复合体的功能一致性
Tab.2 Comparison of function homogeneity of the complexes detected by using different protein networks
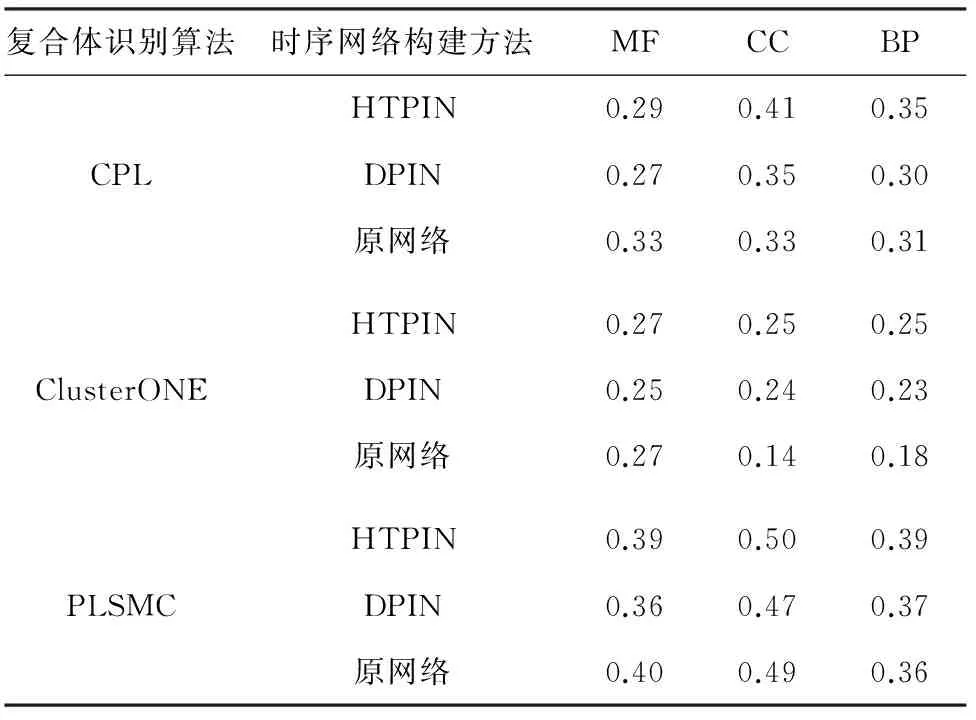
复合体识别算法时序网络构建方法MFCCBPHTPIN0.290.410.35CPLDPIN0.270.350.30原网络0.330.330.31HTPIN0.270.250.25ClusterONEDPIN0.250.240.23原网络0.270.140.18HTPIN0.390.500.39PLSMCDPIN0.360.470.37原网络0.400.490.36
表2列出了CPL、ClusterONE和PLSMC三种算法在不同类型网络上识别复合体的功能一致性比例. 表2列出了关于GO所包含的三个方面功能注释的分析结果,分别为分子功能(MF)、细胞组件(CC)和生物过程(BP). 从表2可以看出,所有参与测试识别算法在HTPIN与DPIN两种时序蛋白质网络上所识别的复合体,在细胞组件和生物过程两个方面的功能一致性均要优于在原始网络上的识别结果. 并且,基于HTPIN网络的识别结果要优于基于DPIN的识别结果. 上述实验结果表明,利用HTPIN方法构建的时序网络可以提高识别复合体的功能一致性.
3 结论与展望
动态-静态蛋白质混合的时序蛋白质网络构建方法的主要特点是,在构建各时刻网络过程中,同时考虑动态蛋白质与静态蛋白质的相互作用关系. 实验结果表明,这种具有混合特征的时序网络能够提高复合体识别的准确性,从而验证了基于“动-静混合”思想构建时序蛋白质网络的方法是合理的.
值得注意的是,本文方法中的波动性阈值本质上是用于控制网络中静态与动态蛋白质比例的一种参数. 文中虽然考察了该参数的不同取值对网络构建的影响,但并未关注该参数的具体计算方法. 作为下一步的工作,将继续研究用于确定波动性阈值的自动计算方法,以增强本文方法的实际应用能力.
[1] TONG A H, DREES B, NARDELLI G, et al. A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules[J]. Science, 2002, 295(5553): 321-324.
[2] SPIRIN V, MIRNY L A. Protein complexes and functional modules in molecular networks[J]. Proceedings of the National Academy of Sciences, 2003, 100(21):12123-12128.
[3]冀俊忠, 刘志军, 刘红欣, 等. 蛋白质相互作用网络功能模块检测的研究综述[J]. 自动化学报, 2014, 40(4):577-593.
JI Junzhong, LIU Zhijun, LIU Hongxin, et al. An overview of research on functional module detection for protein-protein interaction networks[J]. Acta Automatica Sinica, 2014, 40(4): 577-593.
[4]鱼亮, 高琳, 孙鹏岗. 蛋白质网络中复合体和功能模块预测算法研究[J]. 计算机学报, 2011, 34(7):1239-1251.
YU Liang, GAO Lin, SUN Penggang. Research on algorithms for complexes and functional modules prediction in protein-protein interaction networks[J]. Chinese Journal of Computers, 2011,34(7): 1239-1251.
[5]汤希玮, 王建新, 胡秋玲. 蛋白质复合物预测方法分析与比较[J]. 计算机应用研究, 2011, 28(10): 3611-3614.
TANG Xiwei, WANG Jianxin, HU Qiuling. Analysis and compare of methods predicting protein complex[J]. Application Research of Computers[J]. 2011, 28(10): 3611-3614.
[6]黄海滨, 杨路明, 王建新, 等. 基于复合参数的蛋白质网络关键节点识别技术[J]. 自动化学报, 2008, 34(11):1388-1395.
HUANG Haibin, YANG Luming, WANG Jianxin, et al. Identification technique of essential nodes in protein networks based on combined parameters[J]. Acta Automatica Sinica, 2008, 34(11): 1388-1395.
[7] LICHTENBERG U, JENSEN L J, BRUNAK S, et al. Dynamic complex formation during the yeast cell cycle[J]. Science, 2005, 307(5710):724-727.
[8] TANG Xiwei, WANG Jianxin, LIU Binbin, et al. A comparison of the functional modules identified from time course and static PPI network data[J]. BMC Bioinformatics, 2011, 12(1):1-15.
[9] WANG Jianxin, PENG Xiaoqing, PENG Wei, et al. Dynamic protein interaction network construction and applications[J]. Proteomics, 2014, 14(4-5): 338-352.
[10]HEGDE S R, MANIMARAN P, MANDE S C. Dynamic changes in protein functional linkage networks revealed by integration with gene expression data[J]. PLoS Computational Biology, 2008, 4(11):e1000237.
[11]WANG Jianxin, PENG Xiaoqing, LI Min, et al. Construction and application of dynamic protein interaction network based on time course gene expression data[J]. Proteomics, 2013, 13(2):301-312.
[12]KOMUROV K, WHITE M. Revealing static and dynamic modular architecture of the eukaryotic protein interaction network[J]. Molecular Systems Biology, 2007, 3(1):110.
[13]JANJIC V, SHARAN R, PRZULJ N. Modelling the Yeast Interactome[J]. Scientific Reports, 2014, 4:4273.
[14]PU S, WONG J, TURNER B, et al. Up-to-date catalogues of yeast protein complexes[J]. Nucleic Acids Research, 2009, 37(3):825-831.
[15]DAI Qiguo, GUO Maozu, GUO Yingjie, et al. A least square method based model for identifying protein complexes in protein-protein interaction network[J]. Biomed Research International, 2013,2014:720960-720960.
[16]DAI Qiguo, GUO Maozu, LIU Xiaoyan, et al. CPL: Detecting protein complexes by propagating labels on protein-protein interaction network[J]. Journal of Computer Science and Technology, 2014, 29(6): 1083-1093.
[17]NEPUSZ T, YU H, PACCANARO A. Detecting overlapping protein complexes in protein-protein interaction networks[J]. Nature Methods, 2012, 9(5): 471-472.
[18]TU B P, KUDLICKI A, ROWICKA M, et al. Logic of the yeast metabolic cycle: temporal compart-mentalization of cellular processes[J]. Science, 2005, 310(5751):1152-1158.
[19]STARK C, BREITKREUTZ B J, REGULY T, et al. BioGRID: a general repository for interaction datasets[J]. Nucleic Acids Research, 2006, 34(suppl 1):D535-D539.
[20]BOYLE E I, WENG S, GOLLUB J, et al. GO: TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes[J]. Bioinformatics, 2004, 20(18):3710-3715.
(编辑 王小唯 苗秀芝)
A method of constructing temporal protein networks by hybridizing dynamic and static proteins
DAI Qiguo1,2,3, GUO Maozu1, LIU Xiaoyan1, WANG Chunyu1
(1.School of Computer Science and Technology, Harbin Institute of Technology, Harbin 150001, China;2. School of Computer Science and Engineering, Dalian Minzu University, Dalian 116600, Liaoning, China;3.Dalian Key Lab of Digital Technology for National Culture(Dalian Minzu University), Dalian 116600, Liaoning, China)
Public available protein networks at present are static, which could not be used to describe the dynamic characteristics of proteins in a cell effectively. It is necessary to construct temporal protein network by integrating other biological data, which reflects the dynamic activities of proteins. Most of previous methods assume that all proteins are dynamic. However, in addition to dynamic protein, there are many static proteins in the cell. To this end, this paper proposes a new method to construct a temporal protein network both with dynamic and static proteins. In the method, proteins are classified into two types of dynamic and static, and then a protein network is constructed on each time point by both considering the interactions of dynamic and static proteins. Experimental test results show that the temporal protein network constructed by using the proposed method can improve the accuracy of the identification of protein complexes, which verified the reliability of the proposed method.
temporal protein network; protein interaction; gene expression; biological network; protein complex identification
10.11918/j.issn.0367-6234.2016.11.007
2015-05-08
国家自然科学基金(91335112,61571163,61532014,61402132,61271346);中央高校基本科研业务费专项资金(DC201501030)
代启国(1985—),男,讲师; 郭茂祖(1966—),男,教授,博士生导师
郭茂祖,maozuguo@hit.edu.cn
TP391
A
0367-6234(2016)11-0041-06
