Zr和(或)Mn替位掺杂γ-TiAl基合金的延性与电子性质
2016-10-14宋庆功杨宝宝赵俊普秦国顺郭艳蕊胡雪兰
宋庆功,杨宝宝,赵俊普,秦国顺,郭艳蕊,胡雪兰
Zr和(或)Mn替位掺杂-TiAl基合金的延性与电子性质
宋庆功1, 2,杨宝宝1,赵俊普1,秦国顺2,郭艳蕊1,胡雪兰2
(1. 中国民航大学理学院低维材料与技术研究所,天津 300300;2. 中国民航大学中欧航空工程师学院,天津 300300)
采用密度泛函理论计算研究Zr和(或)Mn替位掺杂-TiAl形成的8个合金体系的几何结构、形成能、弹性模量、能带结构和重叠布居数等。结果表明:各个掺杂体系的总能量和原子平均形成能均是负值,表明它们具有较好的能量稳定性、在特定条件下是可以实验制备的。Zr或(和)Mn单(双)替位掺杂可以改善此类-TiAl基合金体系的轴比()和弹性模量比(/),特别是Ti12Al11Zr、Ti12Al11Mn、Ti11MnAl11Zr和Ti11ZrAl11Mn的轴比更接近于1,/接近1.75,预报这4个掺杂体系均具有较好的延性。这为探索改善-TiAl基合金的延性提供了理论依据。根据对典型掺杂体系电子性质和重叠布居数分析,发现Zr和Mn掺杂使体系的共价键强度降低、金属键强度增强,从而提高了面间的可动性,有利于改善合金的延性。
-TiAl基合金;Zr掺杂;Mn掺杂;延性;电子性质;第一性原理
-TiAl基合金具有较低密度、较高的高温强度和抗蠕变性能,同时也具有良好的抗氧化性能,是一种综合性能较好的新型航空航天高温结构材料,一直受到人们的重视[1−3]。但由于其室温拉伸延性、断裂性能和高温抗氧化性能较差等缺点,给实际应用造成了很大困难[4−5]。为了改善-TiAl基合金的性能,科研工作者[6−9]进行了大量理论探索和实验研究,取得了许多进展,其中合金化和微合金化方法能通过改变合金的电子结构、键合类型及强度来改变合金的显微组织结构,进而调节材料的宏观性质而受到更多青睐。例如,杨亮等[10]研究表明,在高Nb-TiAl合金中添加Si元素,可以使导脆相(B2)相体积减小,进而使合金的室温拉伸性能得到改善。而QIU等[11]研究显示Fe和Mo均是相稳定元素,且合金Ti-45Al-3Fe-2Mo的晶粒得到细化,在790 ℃具有良好的延性。王海燕等[12]研究表明Mo可以有效改善-TiAl基合金延性,Mo和Ti原子发生强烈的s-s、p-p、d-d电子相互作用,且随着掺杂浓度的增大,电子相互作用增强,有效地束缚了合金中Ti和Al原子的迁移,有助于提高合金的稳定性和强度。黄宇阳等[13]实验制备了Ti50Al48Zr2合金,并用正电子湮没技术对其进行了研究,结果表明,该合金的延性较纯-TiAl显著改善,并确定Zr占据了Al原子格点。宋庆功等[14−15]计算研究表明,经过Zr合金化,-TiAl基合金的热学性质得到改善;并且Zr替位Al原子掺杂浓度为1.85%~6.25%(摩尔分数)时,对该合金延性改善效果较显著。曲选辉等[16]实验研究表明,添加合金元素Mn,可以促进TiAl合金中的孪生变形,从而使其室温延性得以明显改善。陈律等[17]对Mn替位掺杂-TiAl合金进行了第一性原理平面波赝势方法计算,结果表明,Mn替位Al原子掺杂降低了合金中(001)面内原子间的键合强度,相反增强了相邻层间原子间的键合强度,进而改善了该类合金的室温延性。李燕峰等[18]采用基于密度泛函的赝势平面波法对TiAl系合金的电子结构与成键特征进行了计算研究,结果表明,Mn替位Al原子后,使TiAl3合金中Al—Al共价键减少,并抑制了Al原子p轨道与Ti原子d轨道的杂化,从而改善了该合金的室温延性。
关于-TiAl基合金的实验和理论研究已经取得了许多进展,但Zr和Mn元素双掺杂体系的研究尚未见报道。基于这一背景,选择典型的合金元素Zr和Mn,分别单替位和双替位掺杂-TiAl作为研究对象,采用第一性原理计算方法,研究替位掺杂后合金体系的结构、延性、能量性质和电子性质等,以期对-TiAl基合金的研发提供有价值的理论依据。
1 结构模型和计算方法
1.1 结构模型
纯-TiAl合金属于L10超点阵结构[19]。这种结构可视为由一个纯Ti和一个纯Al的简单四方格子相互套构形成的B2型复式格子,如图1(a)所示。报道的纯-TiAl晶格参量的典型实测结果为0=0=2.8373 Å,0=4.0591 Å[20]。为了使计算研究对象更加接近实验制备的-TiAl基合金样品,本研究构建3×2×2的-TiAl超胞模型,如图1(b)所示。Zr和Mn随机替代Al或Ti原子,构建8个替位掺杂-TiAl基合金体系,如图1(c)~(j)所示。
1.2 计算方法与方案
采用密度泛函理论框架下的平面波赝势方法,选择CASTEP软件包[21],利用高性能计算集群进行第一性原理计算研究与分析。交换关联函数采用广义梯度近似(GGA)条件下的PBE形式;电子与离子实之间的相互作用采用超软赝势描述。平面波截断能选取为360 eV,k点设置为4×3×3。自洽场(SCF)计算时采用了Pulay密度混合法,自洽计算误差设置为1.0×10−6eV,收敛标准设置为:每个原子能量变化值低于5.0×10−6eV,原子作用力低于0.1 eV/nm,应力偏差小于0.02 GPa,位移偏移小于5.0×10−6Å。采用BFGS方法进行各个体系的几何结构优化计算,直至得到收敛的结果。优化得到体系S0的晶格参量为=8.467 Å,=5.633 Å,=8.194 Å,它们与相对应的实验值(30=8.512 Å,20=5.675 Å,20=8.118 Å)较为接近,相对误差均在1.0%以下。这表明所选方案比较适合体系S0,对于各个掺杂体系的研究,均采用这种计算方案。对于各个掺杂体系物理性质的计算研究,均以优化后的晶体几何结构为基础,并采用与该体系的几何优化相同的计算方案来完成。
2 结果与讨论
2.1 形成能和结构稳定性
为讨论便利,分别用S0,S1,…,S8表示纯-TiAl体系(Ti12Al12),单掺杂和双掺杂-TiAl基合金体系,如表1所列。材料的稳定性在一定程度上可以用形成能进行表征。原子平均形成能越低,则该材料的稳定性越好[22]。掺杂-TiAl基合金体系的原子平均形成能b[23]可以表示为

式中:t为晶胞体系的总能量;、、和分别表示晶胞中各种元素的原子数;为晶胞体系的总原子数;Ti、Al、Zr和Mn分别为各原子在单质情况下的单原子能量。采用同样的计算方案和参数设置,计算得到Ti、Al、Zr和Mn的值分别为−1603.0999,−56.3896、−1281.0852和−653.5876 eV。计算得到各个合金体系的总能量t如表1所列。根据式(1)计算出各个体系的原子平均形成能b,结果如表1所列。
从表1可以看出,与纯-TiAl体系相比,各个掺杂体系的原子平均形成能均有所升高,因而稳定性有所降低。对于单掺杂体系,bS2<bS1,表明Zr更倾向于替代Ti原子。这与JIANG等[24]的理论计算结论一致。bS3<bS4,表明Mn更倾向于替代Al原子。这与MOHANDAS等[25]以及SONG等[26]报道的实验结果可以相互佐证。

图1 未掺杂和掺杂γ-TiAl体系的结构模型

表1 γ-TiAl基合金体系的总能量和形成能
Zr和Mn双掺杂时,情况较为复杂。平均形成能的大小关系为bS8<bS7<bS5<bS6,故该4个双掺杂体系的稳定性由大到小依次为S8、S7、S5、S6。这说明Zr和Mn双掺杂时,Zr和Mn均替代Ti原子会有较大的概率发生,进而形成Ti10ZrMnAl12体系。
各个掺杂-TiAl基合金体系的总能量和原子平均形成能均为负值。这表明这些合金体系都具有能量稳定性。这也预示着这些体系在一定条件下是可以实验上制备的,有兴趣的实验研究者可以试制。
2.2 掺杂对几何结构及延性的影响
几何优化后,各个体系的晶格参量如表2所列。
已知Ti、Al、Zr和Mn原子半径的关系为Zr>Ti>Al>Mn[27]。计算结果表明,对于单掺杂体系,杂质Mn原子使体系的晶胞体积变小;而杂质Zr原子则使体系的晶胞体积变大。这与原子半径变化趋势一致,可以说各个体系的晶胞体积的变化直接受到杂质原子半径的影响。对于双掺杂体系,晶胞体积的变化受到较大的Zr原子半径和较小的Mn原子半径的共同制约。计算结果显示,各个双掺杂体系的晶胞体积都有所增大,表明杂质Zr原子的影响为主、杂质Mn原子的影响为辅。

表2 γ-TiAl基合金体系的几何性质
掺杂使合金体系的晶格参量和发生了变化,导致其轴比(是把优化计算所得晶格参量由体心四方转变成等价的L10结构后的轴比)较纯-TiAl体系有所变化。为便于分析,将各个体系的轴比绘于图2中。

图2 γ-TiAl基合金体系的轴比
KAWABATA等[28]研究发现,合金的晶胞轴比在一定程度上可以反应其延性,即轴比越接近于1,合金的延性越好。由表2和图2可知,Zr单掺杂体系S1,S2的轴比均较纯-TiAl的更接近1,即Zr可以有效改善该类合金的延性。这个结论可与黄宇阳等[13]的实验结果和宋庆功等[15]的理论结果均可相互佐证。同理,Mn单掺杂体系S3的延性较纯-TiAl的有所改善,此结果可与陈律等[17]的计算结果相互佐证;相反,S4的延性则较纯-TiAl的有所降低。而Mn倾向于替代Al原子,此时掺杂使延性改善。这与曲选辉等[16]实验研究结果的趋势一致。
与立方晶系对比,掺杂体系S1、S6和S7的轴比的相对误差均小于0.7%。这对于改善合金体系的延性具有明显的指示作用。基于轴比的理论预测为实验制备延性较好的-TiAl基合金提供了依据。
2.3 掺杂对弹性模量及延性的影响
γ-TiAl基合金属于简单四方晶系,其弹性常数有6个独立分量,即11、12、13、33、44和66。对各个稳定体系进行第一性原理计算,得到弹性常数如表3所列。
四方晶系的体弹性模量和剪切弹性模量与弹性常数的关系[29]分别为
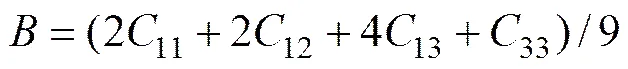
(3)
根据式(2)和(3),计算得到了各个体系的和如表4所列。为便于讨论,也将/和/列于表4中。

表3 γ-TiAl基合金体系的弹性常数

表4 γ-TiAl基合金体系的弹性模量及其比值
由表4可以看出,各个体系的和均发生变化。对单掺杂体系,S3和S4的和均变小;S1的增大,变小;S2的和均增大。对双掺杂体系,S6和S8的和均变小;S5和S7的均略有增大,而明显变小。由于和变化情况各异,因而对体系的延性和塑性产生影响的程度明显不同。
PUGH等[30]报道,依据材料可以衡量材料的延性。即/越大,材料越脆;/越小,材料的延性越好。FU[31]研究指出,上述规律对金属间化合物材料也同样适用。根据表4绘制掺杂-TiAl基合金体系的/关系,如图3所示。对单掺杂体系,当Mn替代Ti原子时,合金体系S4的/略微增加,不利于改善其延性。这与轴比计算的结果吻合。其余各个掺杂体系的/都有所降低,特别是体系S1、S3、S5、S6和S7的/下降明显,对于改善合金体系的延性有利。其中对S1,S3,S6和S7的预测与根据轴比的预测结果一致。
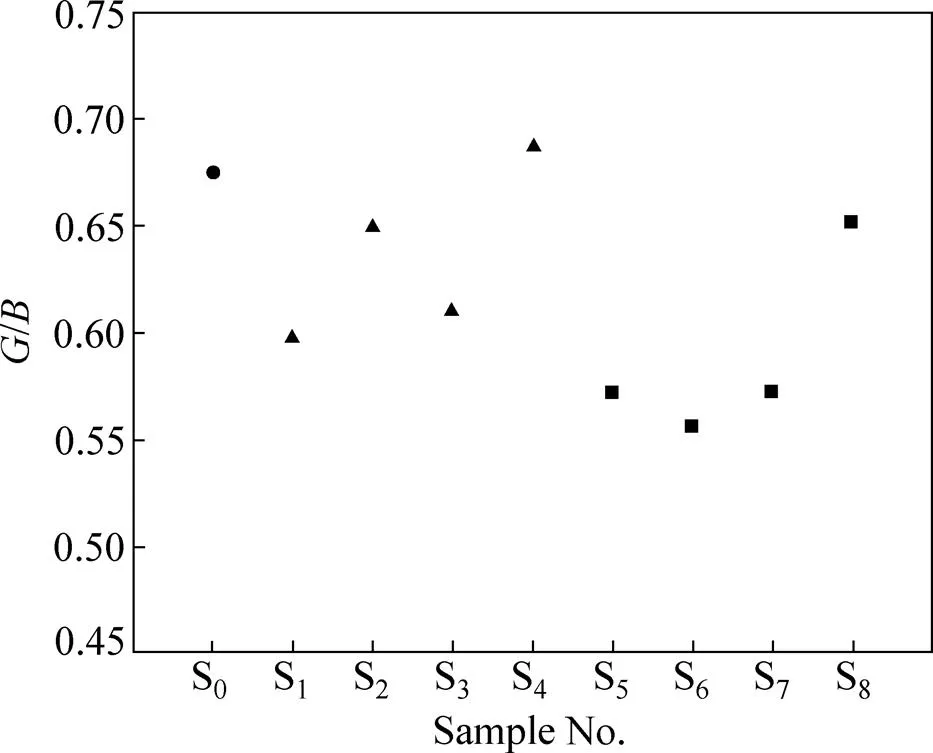
图3 γ-TiAl基合金体系的G/B
材料的延性可通过/来衡量,/≥1.75预示材料延性较好[32]。表4显示体系S5、S6和S7是较好的延性材料。由于S5的轴比太小,其延性尚不能完全定性。
综合对轴比、/和/的分析,可以预测Zr和(或)Mn掺杂γ-TiAl基合金体系S1、S3、S6和S7的延性较好。
2.4 电子性质与结构稳定性
晶体材料的电子性质可以通过其能带结构(Band structure)和态密度(DOS)来分析。本研究仅对轴比最接近于1的3个掺杂体系S1、S6和S7以及纯-TiAl体系S0进行对比分析。计算得到的能带结构如图4所示,其中费米能级位于能量为零处。容易看出,3个掺杂体系的价带能级密集,且价带顶均在费米能级之上。这表明它们都具有典型的电子导电性质,也就是金属导电性。
掺杂-TiAl基合金体系的电子性质与其结构稳定性也密切相关。CARLSSON等[33]和XU等[34]研究表明,就不同结构晶体而言,费米能级处的电子态密度越低,其结构越稳定。本研究计算出上述4个体系的态密度(DOS)和分波态密度(PDOS)如图5所示。
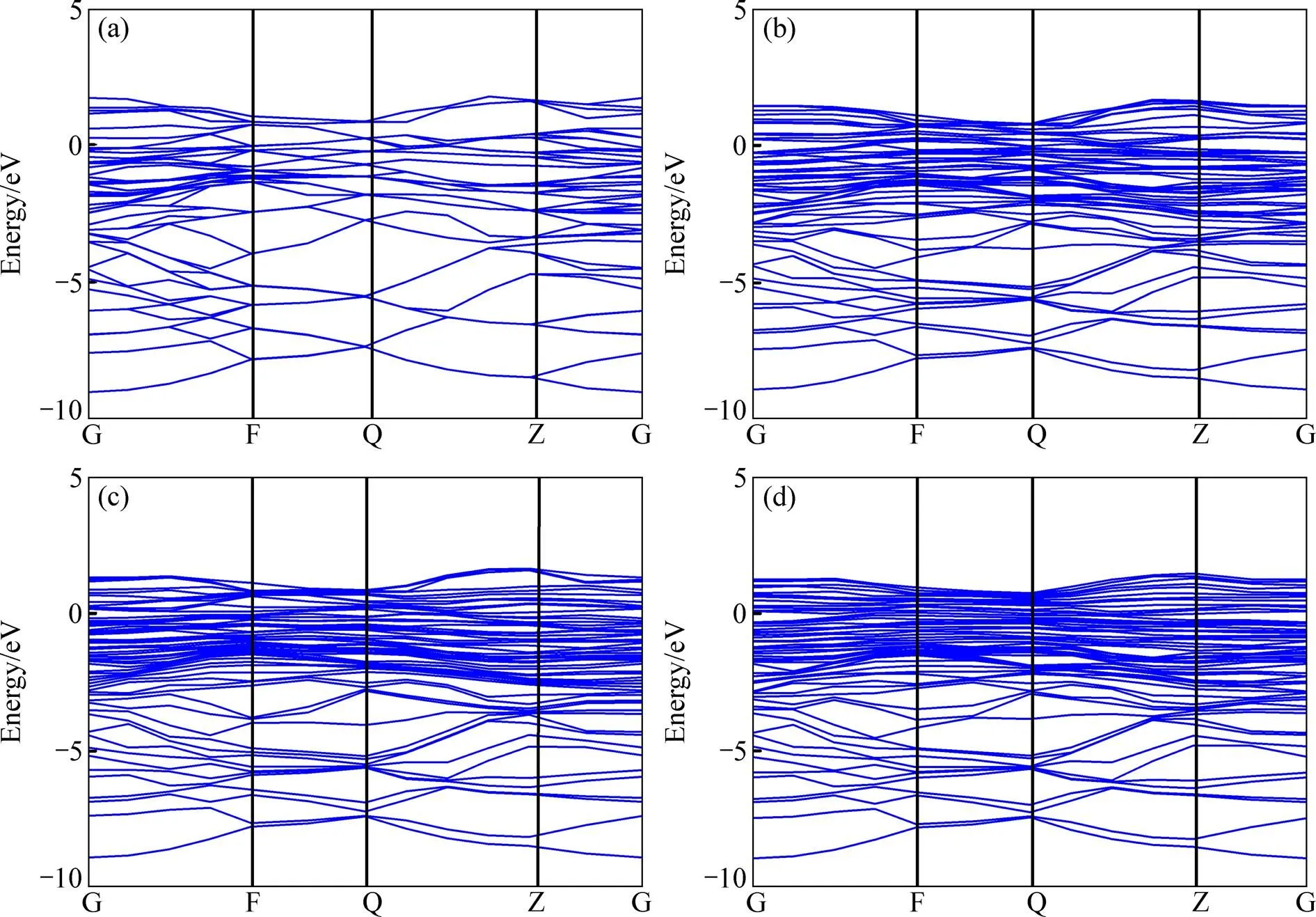
图4 γ-TiAl基合金体系的能带结构
2.5 化学键与延性
掺杂-TiAl基合金体系的金属性质与其中原子间结合的化学键密切相关。本研究计算了Zr单掺杂体系S1和纯-TiAl体系S0中各个原子间的重叠布居数,如表5所列。由表5可以看出,在纯-TiAl 体系S0中,电荷从Ti-4s轨道转移到了Ti-3d轨道,同时也有电荷从Al-3s轨道转移到Al-3p轨道,这直接导致-TiAl体系中p-d轨道杂化作用的加强[35]。在平行于面的同层Al原子之间,它们之间有较大的重叠布居数(0.37~0.65),这表明它们之间有很强的共价结合作用;而Al与其近邻Ti原子之间的重叠布居数仅为0.13,也表明有一定的共价键性质。在Zr单掺杂体系S1中,Zr替代1个Al原子,它与其最近邻的8个Ti原子之间的重叠布居数降低为负值(−0.18),显示Zr与Ti原子之间形成反键,没有共价结合特征;而Zr与其同层Al原子之间的重叠布居数也有所变化,但总的效果是降低的,因而导致共价结合强度有所降低。其他原子间重叠布居数变化不大。结合能带结构的金属特征,推断体系S1中金属键增强,其延性得以改善。
Zr和Mn双掺杂体系S6和S7中原子间的重叠布居数如表6所列。在S6中,Zr与其最近邻的7个Ti原子之间的重叠布居数大幅降低,量值为−0.28~ −0.13。Zr与Mn原子之间重叠布居数为−0.06。这表明,Zr与其最近邻Ti或Mn原子的结合没有共价键性质;而Zr与在同一面的4个Al原子之间有较大的重叠布居数(0.38~0.58),与纯-TiAl差别不大,这表明它们之间有很强的共价结合作用。Mn与其最近邻的7个Al原子之间的重叠布居数也有所下降(0.10~0.16),但变化不大,显示Mn与7个Al原子之间仍有较弱的共价键结合;Mn与在同一面的4个近邻Ti原子之间有较大的重叠布居数(0.21~0.24),且其共价键结合增强,其他原子间重叠布居数变化不大。结合能带结构的金属特征,推断体系S6中金属键增强,其延性得以改善。
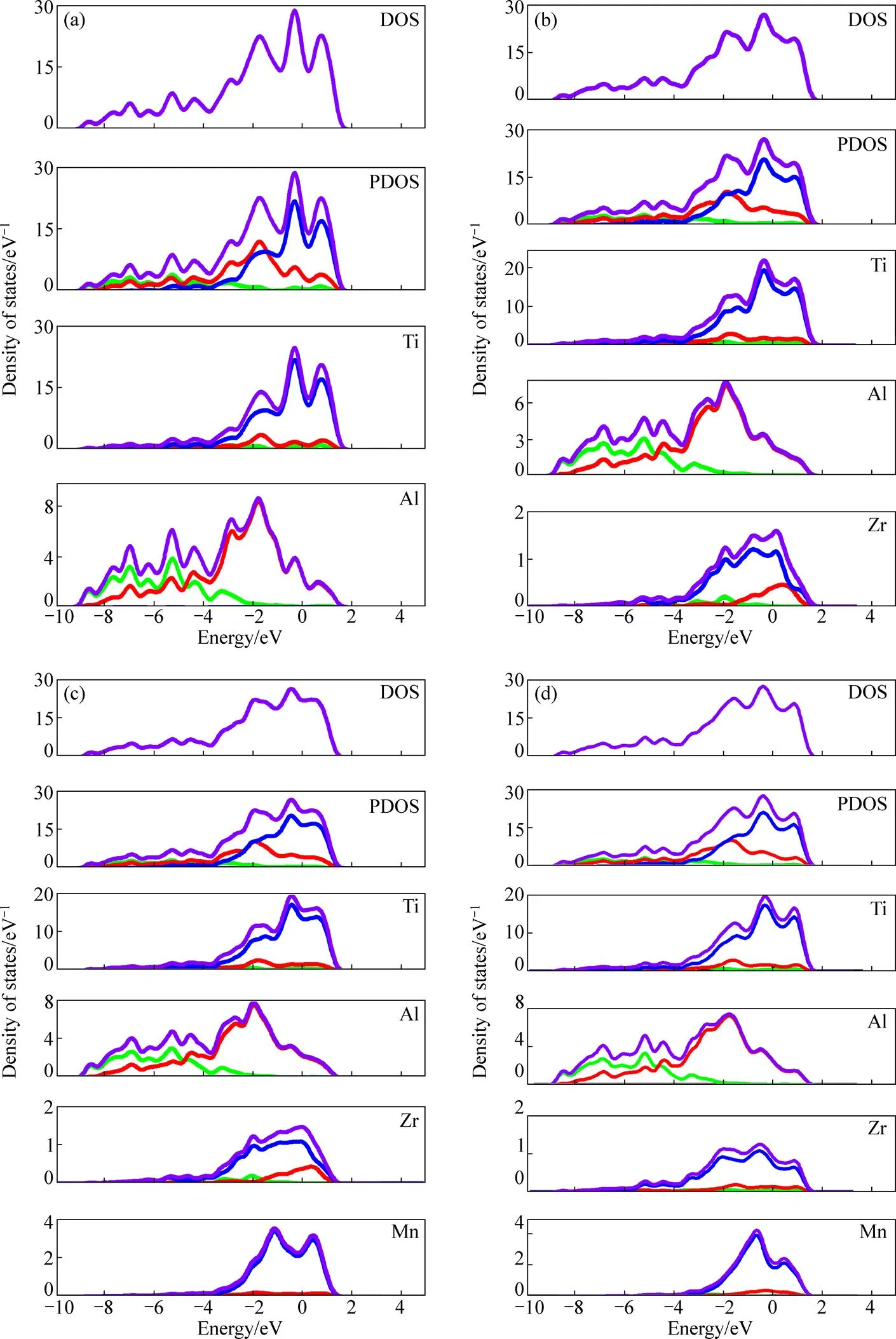
——s;——p;——d;——sum

表5 Zr单掺杂体系S1与纯γ-TiAl体系S0的重叠布居数

表6 Zr和Mn双掺杂γ-TiAl体系S6和S7的重叠布居数
同样,在S7中,Mn与其最近邻的7个Ti原子之间的重叠布居数降至0.06~0.08,Mn与Zr原子之间的重叠布居数降至0,这显示Mn与其最近邻Ti(或Zr)原子间的共价结合强度大大降低。而Mn与在同一面的4个Al原子之间重叠布居数有所下降,但仍然保持较大的量值(0.36~0.51),这表明它们之间仍有很强的共价结合作用;Zr与其最近邻的7个Al原子之间的重叠布居数也降至0.05~0.11,共价键结合强度减弱;而Zr与在同一面的4个近邻Ti原子之间重叠布居数降至−0.14~0.05,共价键结合强度显著降低。其它原子间重叠布居数变化不大。综合而言,原子间重叠布居数大为降低,共价结合强度下降。由于其能带结构的金属特征,推断体系S7中金属键增强,其延性得以改善。
综上所述,在该类金属间化合物中进行替位掺杂Zr或(和)Mn原子,能使合金中的共价键强度降低、金属键结合趋势增大,体系的稳定性降低,提高了晶面间的可动性,这对于合金延性的改善是有利的。这些预测结果与对轴比、弹性模量比的分析结果是一致的。
3 结论
1) Zr或(和)Mn单(双)替位掺杂可以改善此类-TiA基合金体系的轴比,特别是体系S1(Ti12Al11Zr),S3(Ti12Al11Mn),S6(Ti11MnAl11Zr)和S7(Ti11ZrAl11Mn)的轴比都有明显改善,更接近于1。单掺杂情况下,晶胞体积的变化与杂质原子半径的变化趋势一致。双掺杂情况下,晶胞体积都有所增大。这种变化表明晶胞体积变化受两种杂质原子的共同制约,但以Zr原子的影响为主、Mn原子的影响为辅。
2) 各个掺杂体系的总能量和原子平均形成能均是负值,表明它们具有较好的能量稳定性、在实验上是可以制备的。单掺杂情况下,Mn倾向于替代Al原子,而Zr则更倾向于替代Ti原子,与报道实验研究可以相互佐证;双掺杂情况下,Zr仍倾向于替代Ti原子,而Mn既能替代Ti原子,也能替代Al原子。
3) 轴比、弹性模量比均显示体系S1,S3,S6和S7具有较好的延性。这为探索改善-TiAl基合金的延性提供了理论依据,揭示了方向。
4) 能带结构和态密度显示,3个典型掺杂体系S1,S6和S7都具有典型的金属导电性。经过对原子间重叠布居数的计算与分析,发现用Zr或(和)Mn原子对金属间化合物-TiAl进行替位掺杂,能使其中的共价键强度降低、金属键结合趋势增强,提高了晶面间的可动性,这对于合金延性的改善是有利的。
REFERENCES
[1] KUNAL K, RAMACHANDRAN R, NORMAN M. Advances in gamma titanium aluminides and their manufacturing techniques[J]. Progress in Aerospace Sciences, 2012, 55(12): 1−16.
[2] CLEMENS H, MAYER S. Design, processing, microstructure, properties, and applications of advanced intermetallic TiAl alloys[J]. Advanced Engineering Materials, 2013, 15(4): 191−215.
[3] WANG Ji-qiang, KONG Ling-yan, LI Tie-fan, XIONG Tian-ying. High temperature oxidation behavior of Ti(Al, Si)3diffusion coating on-TiAl by cold spray[J]. Transactions of Nonferrous Metals Society of China, 2016, 26(4): 1155−1162.
[4] WU X H. Review of alloy and process development of TiAl alloys [J]. Intermetallics, 2006, 6(10/11): 14−22.
[5] 陶辉锦, 彭 坤, 谢佑卿, 贺跃辉, 尹志民. Ti-Al 金属间化合物脆性问题的研究进展[J]. 粉末冶金材料科学与工程, 2007, 12(6): 330−336. TAO Hui-jin, PENG Kun, XIE You-qing, HE Yue-hui, YIN Zhi-min. Research advance on brittleness of Ti-Al intermetallics[J]. Materials Science and Engineering of Powder Metallurgy, 2007, 12(6): 330−336.
[6] KARTAVYKH A V, ASNIS E A, PISKUN N V, STATKEVICH I I, GORSHENKOV M V. Microstructure and mechanical properties control of-TiAl(Nb, Cr, Zr) intermetallic alloy by induction float zone processing[J]. Journal of Alloys and Compounds, 2015, 643(S1): S182−S186.
[7] IRNAYEV R M, IMAYEV V M, OEHRING M. Alloy design concepts for refined gamma titanium aluminide based alloys[J]. Intermetallics, 2007, 15(4): 451−460.
[8] 肖代红, 黄伯云. 铸造Ti-47Al-8Cr-2Nb合金的低温超塑性及其组织演变[J]. 中国有色金属学报, 2008, 18(10): 1750−1755.XIAO Dai-hong, HUANG Bai-yun. Superplastic behavior and microstructure evolution of as-cast Ti-47Al-8Cr-2Nb alloy at lower temperature [J]. The Chinese Journal of Nonferrous Metals, 2008, 18(10): 1750−1755.
[9] 苏继龙, 连兴峰.-TiAl 基合金弹性和塑性尺度效应所对应内在特征尺寸的关系[J]. 中国有色金属学报, 2015, 25(2): 338−343. SU Ji-long, LIAN Xing-feng. Relationship between intrinsic characteristic sizes of elastic property and plastic property of-TiAl based alloy[J]. The Chinese Journal of Nonferrous Metals, 2015, 25(2): 338−343.
[10] 杨 亮, 高叔博, 王艳丽, 叶 腾, 宋 霖, 林均品. Si对高Nb-TiAl合金组织及室温拉伸性能的影响[J]. 金属学报, 2015, 51(7): 859−865. YANG Liang, GAO Shu-bo, WANG Yan-li, YE Teng, SONG Lin, LIN Jun-pin. Effect of Si addition on the microstructure and room temperature tensile properties of high Nb-TiAl alloy[J]. Acta Metallurgica Sinica, 2015, 51(7): 859−865.
[11] QIU C Z, LIU Y, HUANG L, ZHANG W, LIU B, LU B. Effect of Fe and Mo additions on microstructure and mechanical properties of TiAl intermetallics[J]. Transactions of Nonferrous Metals Society of China, 2012, 22(3): 521−527.
[12] 王海燕, 胡前库, 杨文朋, 李旭升. 金属元素掺杂对TiAl合金力学性能的影响[J]. 物理学报, 2016, 65(7): 1−9. WANG Hai-yan, HU Qian-ku, YANG Wen-peng, LI Xu-sheng. Influence of metal element doping on the mechanical properties of TiAl alloy[J]. Acta Physica Sinica, 2016, 65(7): 1−9.
[13] 黄宇阳, 吴伟明, 邓 文, 钟夏平, 熊良钺, 曹名洲, 龙期威. 用正电子湮没技术研究Zr和Nb在TiAl合金中的行为[J]. 中国有色金属学报, 2000, 10(6): 796−799. HUANG Yu-yang, WU Wei-ming, DENG Wen, ZHONG Xia-ping, XIONG Liang-yue, CAO Ming-zhou, LONG Qi-wei. Behavior of Zr and Nb in TiAl alloy investigated by positron annihilation technique[J]. The Chinese Journal of Nonferrous Metals, 2000, 10(6): 796−799.
[14] 宋庆功, 闫洪洋, 果福娟, 徐霆耀, 康建海, 胡雪兰. Zr替位掺杂-TiAl的稳定性和热学性质研究[J]. 功能材料. 2014, 45(19): 19149−19154. SONG Qing-gong, YAN Hong-yang, GUO Fu-juan, XU Ting-yao, KANG Jian-hai, HU Xue-lan. An investigation on the stability and thermal property of-TiAl doped with Zr-substitution[J]. Journal of Functional Materials, 2014, 45(19): 19149−19154.
[15] 宋庆功, 秦国顺, 杨宝宝, 蒋清杰, 胡雪兰. 杂质浓度对Zr替位掺杂-TiAl合金的结构延性和电子性质的影响[J]. 物理学报. 2016, 65(4): 244−252. SONG Qing-gong, QIN Guo-sun, YANG Bao-bao, JIANG Qing-jie, HU Xue-lan. Impurity concentration effects on the structures, ductile and electronic properties of Zr-doped gamma-TiAl alloys[J]. Acta Physica Sinica, 2016, 65(4): 244−252.
[16] 曲选辉, 黄伯云, 吕海波, 黄培云, 孔祥炎, 钱 沅. 添加锰对TiAl金属间化合物孪生变形的影响[J]. 中南矿冶学院学报, 1992, 23(3): 296−302. QU Xuan-hui, HUANG Bai-yun, LÜ Hai-bo, HUANG Pei-yun, KONG Xiang-yan, QIAN Yuan. Effects of Mn-addition on twinning deformation in TiAl intermetallics[J]. Journal of Central South University, 1992, 23(3): 296−302.
[17] 陈 律, 彭 平, 李贵发, 胡艳军, 周惦武, 张为民. L10-TiA l金属间化合物Mn, Nb合金化电子结构的计算[J]. 航空材料学报, 2005, 25(5): 15−19. CHEN Lü, PENG Ping, LI Gui-fa, HU Yan-jun, ZHOU Dian-wu, ZHANG Wei-min. First-principles study on electron ic structure of L10-TiAl intermetallic compound alloyed by Mn or Nb[J]. Journal of Aeronautical Materials, 2005, 25(5): 15−19.
[18] 李燕峰, 徐 慧, 夏庆林, 宋招权. Ti-Al系电子结构及Mn掺杂对TiAl3室温脆性的影响[J]. 粉末冶金材料科学与工程, 2010, 15(2): 102−109. LI Yan-feng, XU Hui, XIA Qing-lin, SONG Zhao-quan. Influence of Ti-Al electronic structure and Mn-doping brittleness TiAl3at room temperature[J]. Materials Science and Engineering of Powder Metallurgy, 2010, 15(2): 102−109.
[19] 黄伯云. 钛铝基金属间化合物[M]. 长沙:中南工业大学出版社, 1998. HUANG Bai-yun. Intermetallics of titanium aluminides[M]. Changsha: Central South University of Technology Press, 1998.
[20] NOVOSELOVA T, MALINOW S, SHA W, ZHECHEVA L. High-temperature synchrotron X-ray diffraction study of phases in a gamma TiAl alloy[J]. Materials Science and Engineering A, 2004, 371(1/2): 103−112.
[21] SEGALL M D, LINDAN P J D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE, M C. First-principles simulation: Ideas, illustrations and the CASTEP code[J]. Journal of Physics: Condensed Matter, 2002, 14(11): 2717−2744.
[22] 宋庆功, 姜恩永. 快离子导体AgTiS2中Ag+离子−空位的二维基态结构与能量性质研究[J]. 物理学报, 2008, 57(3): 1823−1828. SONG Qing-gong, JIANG En-yong. Study on the structural andenergetic properties of two-dimensional ground state of Ag+ion-vacancy in fast ionic conductor AgTiS2[J]. Acta Physica Sinica, 2008, 57(3): 1823−1828.
[23] KONG F T, CHEN Z Y, TIAN J, CHEN Y Y. Methods of improving room temperature ductility of TiAl based alloys[J]. Rare Metal Materials and Engineering, 2003, 32(2): 81−86.
[24] JIANG C. First-principles study of site occupancy of dilute 3d, 4d and 5d transition metal solutes in L10TiAl[J]. Acta Materialia, 2008, 56(20): 6224−6231.
[25] MOHANDAS E, BEAVEN P A. Site occupation of Nb, V, Mn and Cr in-TiAl[J]. Scripta Metallurgica et Materialia, 1991, 25(9): 2023−2027.
[26] SONG Y, YANG R, LI D, HU Z Q, GUO Z X. A first principles study of the influence of alloying elements on TiAl: Site preference[J]. Intermetallics, 2000, 8(5): 563−568.
[27] SPEIGHT J G. Lange’shandbook ofchemistry16th ed[M]. Boston: McGraw-Hill Professional, 2005.
[28] KAWABATA T, TAMURA T, IZUMI O. Effect of Ti/Al ratio and Cr, Nb, and Hf additions on material factors and mechanical properties in TiAl[J]. Metallurgical and Materials Transactions A, 1993, 24(1): 141−150.
[29] PABST W, GREGOROVA E. Effective elastic properties of alumina-zirconia composite ceramics-part 2. Micromechanical modeling[J]. Ceramics-Silikaty, 2004, 48(1): 14−23.
[30] PUGH S F. Relation between the elastic moduli and the plastic properties of polycrystalline pure metals[J]. Philosophical Magazine, 1954, 45(367): 823−843.
[31] FU C L. Electronic, elastic, and fracture properties of trialuminide alloys: Al3Sc and Al3Ti[J]. Journal of Materials Research, 1990, 5(5): 971−979.
[32] WANG Q Q, ZHAO Z S, XU L F, WANG L M, YU D L, TIAN Y J, HE J L. Novel high-pressure phase of RhB: First-principle calculations[J]. The Journal of Physical Chemistry C, 2011, 115(40): 19910−19915.
[33] CARLSSON A E, MESCHTER P J. Relative stability of L12, DO22, and DO23structures in MAl3 compounds[J]. Journal of Materials Research, 1989, 4(5): 1060−1063.
[34] XU J H, FREEMAN A J. Band filling and structural stability of cubic trialuminides: YAl3, ZrAl3, and NbAl3[J]. Physical Review Series B, 1989, 40(17): 11927−11930.
[35] 党宏丽, 王崇愚, 于 涛.-TiAl中Nb和Mo合金化效应的第一性原理研究[J]. 物理学报, 2007, 56(5): 2838−2844. DANG Hong-li, WANG Chong-yu, YU Tao. First-principles investigation on alloying effect of Nb and Mo in-TiAl[J]. Acta Physica Sinica, 2007, 56(5): 2838−2844.
(编辑 王 超)
Investigations on ductibility and electronic property of Zr and (or) Mn doped-TiAl based alloys
SONG Qing-gong1, 2, YANG Bao-bao1, ZHAO Jun-pu1, QIN Guo-shun2, GUO Yan-rui1, HU Xue-lan2
(1. Institute of Low Dimensional Materials and Technology, College of Science,Civil Aviation University of China, Tianjin 300300, China;2. Sino-European Institute of Aviation Engineering, Civil Aviation University of China, Tianjin 300300, China)
8 kinds of-TiAl based alloys doped with Zr and (or) Mn were investigated with density function theory. The geometric structures, formation energies, elasticity modulus, band structures and overlap populations were analyzed. The results indicate that these systems possess energy stability and can be made with experiment at certain condition because of their negative total energies and atomic average formation energies. Zr and (or) Mn doping can change the axis ratios () and elastic moduli ratios (/) of the 8 systems. It is forecasted that systems of Ti11MnAl11Zr, Ti12Al11Zr, Ti12Al11Mn, and Ti11ZrAl11Mn possess good ductibility for their axis ratios are close to 1 and moduli ratios are close to 1.75, which provides theoretical support for improving ductibility of-TiAl based alloys. Based on the obtained electronic properties and overlap population, it is argued that the doping of Zr and (or) Mn can make the decrease of covalent bond intension and the increase of metal bond intension, resulting in the improving mobility of the crystal face, which is propitious to the improvement of the intermetallic ductility.
-TiAl based alloy; Zr doping; Mn doping; ductibility; electronic property; first-principles
Project(5120118) supported by the National Natural Science Foundation of China; Project(3122015K001)supported by the Fundamental Research Fundsforthe Central Universities, China
2015-11-23; Accepted date:2016-07-03
SONG Qing-gong; Tel: +86-22-24092510; E-mail: qgsong@cauc.edu.cn
1004-0609(2016)-11-2309-10
TG146.2
A
国家自然科学基金资助项目(51201181);中国民航大学中央高校基本科研业务费资助项目(3122015K001)
2015-11-23;
2016-07-03
宋庆功,教授,博士;电话:022-24092510;E-mail:qgsong@cauc.edu.cn
