分阶段加料熔融法制备Fe1-xO基氨合成催化剂的研究
2016-03-25黄仕良程田红韩文锋刘化章
黄仕良,程田红,韩文锋,刘化章
(浙江工业大学工业催化研究所 浙江杭州 310032)
分阶段加料熔融法制备Fe1-xO基氨合成催化剂的研究
黄仕良,程田红,韩文锋,刘化章
(浙江工业大学工业催化研究所 浙江杭州 310032)
采用熔融法制备Fe1-xO基氨合成催化剂,根据氧化物助催化剂的熔点、晶型和酸碱性等性质,考察了催化剂制备过程中助催化剂按顺序分阶段加料的方式对催化剂活性及热稳定性的影响。结果表明,助催化剂的添加顺序对催化剂氨合成活性有一定的影响;助催化剂按其酸碱性顺序加入对催化剂氨合成活性有一定的影响,其中先加碱性助催化剂制备的催化剂的耐热稳定性高于其他方法制备的催化剂;助催化剂钒和钨以其钙盐形式引入比以钾盐形式引入所得催化剂的耐热活性高。
熔融法 氨合成 Fe1-xO基催化剂 制备方法
氨合成熔铁催化剂的制备曾提出过多种方法,如拜尔法[1]、烧结法[2- 3]、沉淀法[4- 6]、浸渍法[7- 9]等,但工业催化剂的制备依然采用了熔融法[10]。
熔铁催化剂是各种催化剂中制备方法最简单的一种,其制备过程实际上就是“锅煮”式,即将所有的原料全部混合均匀后一次性加入电熔炉[11- 12],除了生成的气体挥发逸出外,所有的固体物料都将留在最终产品中,只要确保加入的生产原料的数量和质量,尤其是原料的纯度,并用零价铁粉调节Fe2+/Fe3+即可满足要求。此过程看似非常简单,但却包含着众多的化学反应、熔融与凝固、结晶与相变、晶粒生长和溶质再分布、偏析等物理化学过程。特别是Fe1-xO基催化剂的发现,由于其在制备原理、化学组成、晶体结构、化学物理性质等方面与传统的Fe3O4催化剂完全不同,因此提出了新的科学问题。Fe1-xO基熔铁催化剂是我国独创的、拥有自主知识产权的创新成果,应该有一个新的理论予以支撑。
在理论研究中发现[13- 15]:①熔铁催化剂表面存在酸碱协同效应;②根据助催化剂与Fe1-xO化学物理结构匹配原则能实现结构掺杂,掺杂合适的氧化物并结合高温快速凝固技术可有效抑制Fe1-xO的岐化反应;③H2的强化学吸附现象与催化剂表面酸碱覆盖度有关;④CaO是主要结构助催化剂,而Al2O3虽然不是主要结构助催化剂,但起着表面重构作用。
关于熔融法制备氨合成催化剂的制备工艺,如熔融温度、熔融时间以及熔浆冷却速率等制备条件对催化剂性能的影响,国内外学者做了大量研究。试验研究表明[10],随着熔融时间的延长,Fe2+/Fe3+呈现先增大后减小的趋势,故需严格控制熔融时间来制备合适Fe2+/Fe3+的催化剂。Lendzion- Bieluń等[16- 17]在研究熔融法制备催化剂过程中,发现助催化剂在催化剂中的分布与熔浆冷却速率有很大关系,冷却速率越慢,催化剂中Al2O3的分布越不均匀,得到的催化剂热稳定性差。本文提出了通过助催化剂按碱性到酸性顺序分阶段加料的方式来改进“锅煮”式熔融法制备氨合成催化剂,以期通过考察助催化剂氧化物的熔点、晶型和酸碱性来研究其分阶段加入次序对催化剂氨合成活性的影响,并考察助催化剂氧化物前驱体对催化剂性能的影响。
1 试验部分
1.1 催化剂制备
根据氧化物助催化剂的酸碱性、熔点以及晶型等物化性质的不同,将物料分批加入电熔炉,一部分助催化剂与精选磁铁矿粉、还原铁粉混合均匀后先加入电熔炉中熔融,待达到熔融状态后再将另一部分助催化剂加入。电熔炉电源由50 kV·A盐浴炉变压器供给,熔融最大电流达2 700 A,最高熔融温度为1 600 ℃。熔融一段时间后,将高温熔浆迅速倾注入带水夹套的冷却槽中冷却至室温,再经破碎、筛分至所需粒度。
1.2 催化剂活性评价
催化剂活性评价在内径为Φ14 mm的连续流动的高压固定床反应器中进行,催化剂粒度为1.0~1.4 mm,并将催化剂装填在反应器等温区内。氢氮混合气(体积比3∶1)由液氨分解制得,经净化除去未分解的NH3,H2O,CO及CO2等杂质气体,净化后的气体采用压缩机升压。催化剂在氢氮混合气压力为5 MPa、空速30 000 h-1和温度400~500 ℃条件下还原28 h,然后在15 MPa、空速30 000 h-1条件下测定反应器出口气体中氨体积分数,再升温至500 ℃恒温16 h,然后在相同条件下重新测定催化剂活性,并考察催化剂的耐热稳定性。
1.3 催化剂表征
NH3- TPD表征:样品量0.5 g,粒度为150~250 μm(100~60目),样品在流量30 mL/min、温度110 ℃的Ar气氛下处理1 h,除去其中的水分;然后降温至50 ℃,以流量30 mL/min通入含氨体积分数为10%的氨氦混合气,恒温0.5 h;切换Ar吹扫1 h;以10 ℃/min升温至800 ℃。
XRD表征:催化剂还原前后的物相用瑞士Thermo ARL X′TRA型X射线衍射仪测定,Cu Kα光源(λ=1.540 56×10-10m),电压45 kV,电流40 mA,扫描速度0.20°/min。
2 试验结果与分析
2.1 按氧化物的熔点和晶型次序添加对催化剂活性的影响
磁铁矿的熔融温度在1 550~1 600 ℃,随着FeO的生成,炉温将逐渐降低,直至稳定在FeO的熔点1 400 ℃左右。助催化剂Al2O3,CaO,K2O等的熔点虽然高于1 600 ℃,但当熔融物处于液体状态时,其均以离子状态存在,理论上应该是均匀分布的,而熔融温度、熔融时间等因素只能影响熔融物处于液体状态时助催化剂的分布状态。一旦熔浆离开电熔炉而发生凝固,则助催化剂在催化剂中的分布状态就基本上被确定,即在凝固过程中会发生溶质再分布而产生偏析,造成分布不均匀。为了使这些助催化剂在催化剂中分散得更均匀,改变助催化剂的添加次序可以减少偏析,如:助催化剂与磁铁矿粉、还原铁粉一起加入炉中,则可延长助催化剂在电熔炉中的扩散时间;而助催化剂V2O5,WO3,MoO3等在高温下都存在显著的升华现象,故在催化剂制备的第2阶段加入,尽量缩短其在电熔炉中的时间,以减少因升华而造成的损失。
Al2O3中的Al3+能够与Fe3+发生阳离子取代[18- 22]而形成取代型固溶体,同时也能与FeO形成化合物型固溶体;Mg2+不仅能够取代Fe2+进入Fe1-xO晶格中,而且MgO可在0%~100%范围内与FeO形成完全固溶体,能均匀地分布在催化剂母体中;CaO可与FeO形成替换固溶体[21]。因此,Al2O3,CaO及MgO都能与FeO形成固溶体,能在催化剂中很好地分散,添加次序不会影响其在催化剂中的分布情况,但考虑到其均是高熔点氧化物,故先投料会更有利,这也与笔者前期的试验结果一致。MnO,CoO及FeO同为立方晶系,只有在高温下容易形成固溶体,为了使其在催化剂中均匀分布,应在制备催化剂的第2阶段加入,即在完全生成FeO的状态下添加MnO或CoO。
因为根据助催化剂氧化物的熔点和晶型确定助催化剂的加入顺序,所以本试验主要考察WO3分别与CoO和MnO作为二次添加和一次性加入法制备的催化剂的氨合成活性进行了比较,催化剂耐热前后活性随温度变化如图1所示,各催化剂的XRD示意如图2所示。
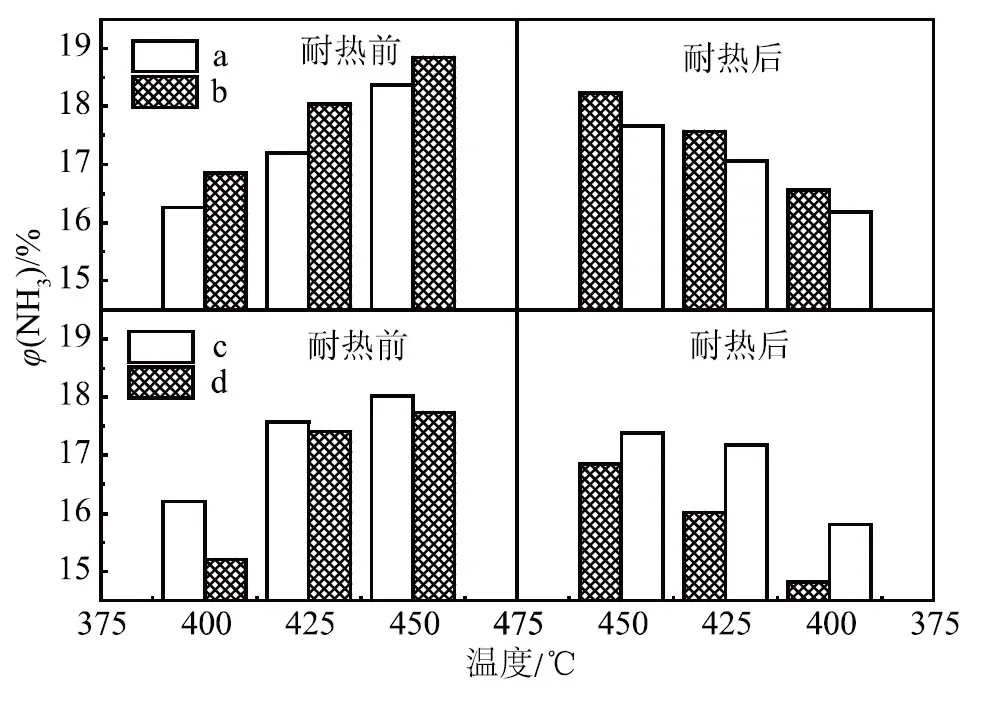
a. 一次性添加 b. 分次加入(WO3和CoO后加)c. 一次性添加 d. 分次加入(WO3和MnO后加)图1 催化剂耐热前后活性随温度变化
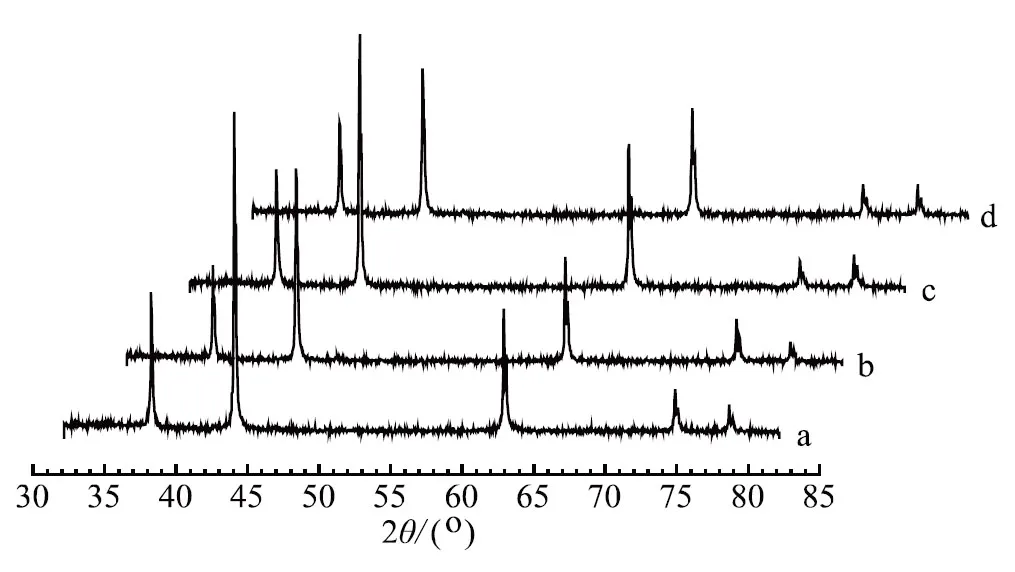
a. 一次性添加 b. 分次加入(WO3和CoO后加)c. 一次性添加 d. 分次加入(WO3和MnO后加)图2 各催化剂的XRD示意
由图1可知:助催化剂WO3和CoO后加所得催化剂耐热前后的活性都高于一次性添加所得催化剂的活性;而助催化剂WO3和MnO的试验结果完全相反,即WO3和MnO一次性添加所得催化剂耐热前后的活性都高于分次加入所得催化剂的活性,在450,425和400 ℃这3个温度点的氨合成活性均是如此。由此可见,添加助催化剂的顺序对氨合成催化剂的活性具有一定影响。
由图2可知,4个样品的XRD图谱中只有FeO相,并未检测到其他物相,其主要原因是添加的助催化剂量少且在催化剂中分散均匀。采用谢乐公式计算FeO不同晶面的晶粒度如表1所示,4个样品FeO(111)晶粒度基本一致,但FeO的另外2个主峰晶粒度差异很大,这意味着在制备催化剂过程中FeO的各晶面生长各向异性,是导致催化剂的氨合成活性不同的可能原因之一。
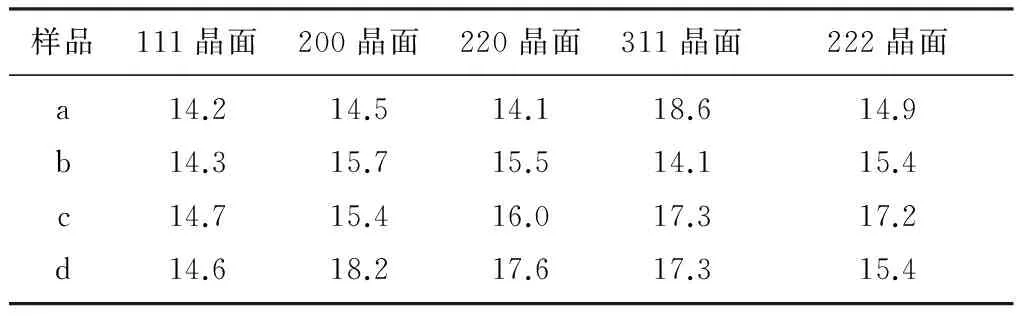
表1 FeO不同晶面的晶粒度 nm
2.2 氧化物的酸碱性对加入次序及催化剂活性的影响
刘化章等[14]在Fe1-xO基催化剂的研究中提出了催化剂表面酸碱协同作用理论,各种助催化剂中,酸性金属氧化物和碱性金属氧化物的含量具有特定的比例,但与其绝对量无关。据此原理,调节酸性金属氧化物和碱性金属氧化物的含量,使催化剂表面酸覆盖度与碱覆盖度的比例为最适宜值时,可提高催化剂的活性。通过BET测定催化剂酸性比表面积和碱性比表面积,并与氨合成活性进行关联,发现当催化剂表面酸碱覆盖度的比例为1.1~1.2时,催化剂具有最高的活性。
Almquist等[23]的研究结果显示,对熔铁催化剂综合利用碱性和酸性氧化物更为有效,并且两者的比例因制备方法不同而有不同的影响。Krabetz等[24]研究发现,催化剂的活性与表面的碱度有一定的关系。刘化章等[25]通过BET表征催化剂酸性比表面积、碱性比表面积与氨合成活性的关系,发现当催化剂表面酸碱覆盖度的比例为1.1~1.2时,催化剂具有最高的活性。然而,催化剂表面既要有利于N2的吸附,又要有利于NH3的脱附,因此,调节催化剂表面的酸碱性十分必要。
显然,这种表面酸碱协同作用与铁氧化物前驱体和催化剂制备方法有关。因此,在铁氧化物前驱体为Fe1-xO的前提下,考察了制备方法对调节催化剂表面酸碱性的影响。根据助催化剂的酸碱性不同,以3种方法加入助催化剂(第1种是不分酸碱性一次性加入助催化剂,第2种是先加酸性助催化剂后加碱性助催化剂,第3种是先加碱性助催化剂后加酸性助催化剂),以考察其对催化剂氨合成活性的影响,试验结果如图3所示。
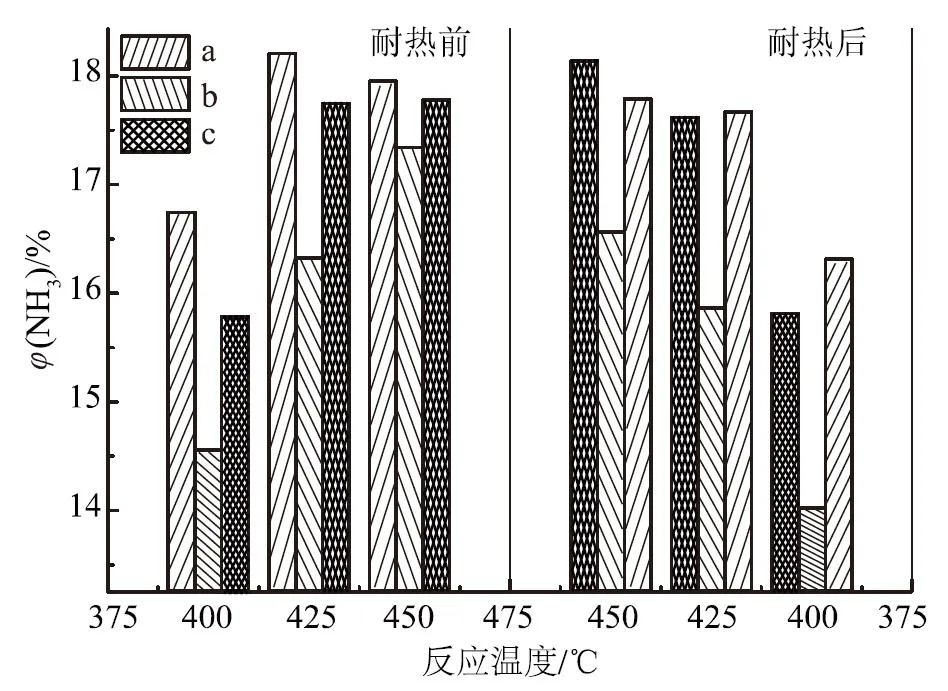
a. 助催化剂一次性加入 b. 分次加入(先加酸性助催化剂) c. 分次加入(先加碱性助催化剂)图3 助催化剂酸碱性及其加料方式对催化剂氨合成活性的影响
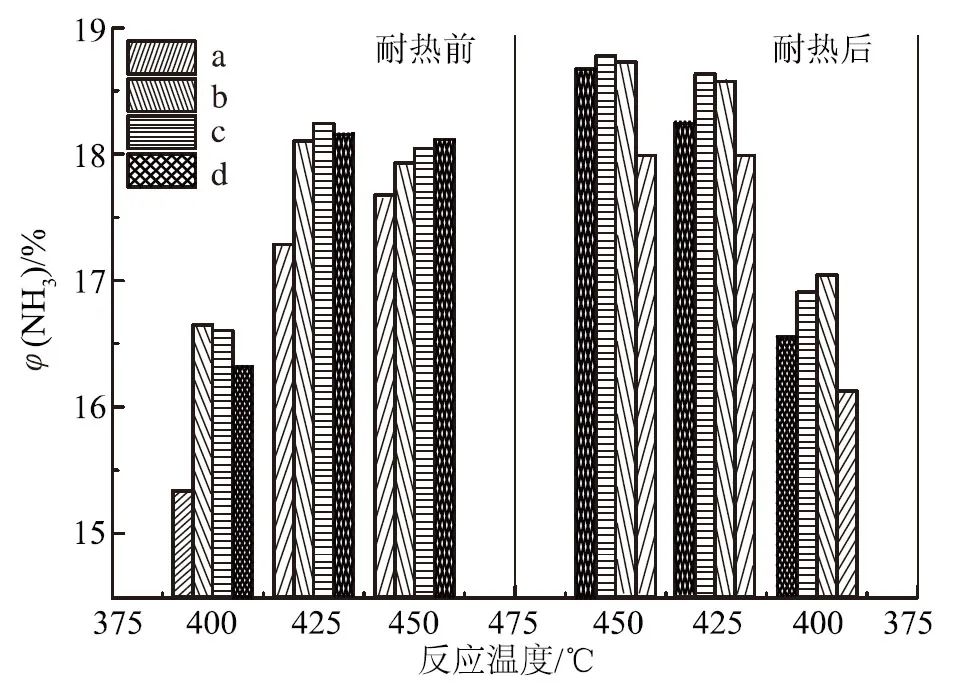
由图3可知,助催化剂的酸碱加入顺序对催化剂氨合成活性有一定影响。在425 ℃耐热前,助催化剂一次性加入的催化剂活性比先加碱性助催化剂的高0.45%,比先加酸性助催化剂的高1.87%,说明在催化剂制备过程中,所有助催化剂一次性加入所得的催化剂活性最好,先加碱性助催化剂次之,先加酸性助催化剂的活性最差。比较耐热前后催化剂氨合成活性可知,在425 ℃时,助催化剂一次性加入的催化剂氨合成活性由耐热前的18.20%降至耐热后的17.66%,绝对值下降0.54%;耐热后,先加酸性助催化剂的催化剂活性绝对值下降0.57%;但先加碱性助催化剂的催化剂活性由耐热前的17.75%降至17.61%,即耐热前后活性基本不变,说明先加碱性助催化剂能提高催化剂耐热稳定性。由此可知,对耐热前的催化剂初活性,助催化剂一次性加入>先加碱性助催化剂>先加酸性助催化剂,助催化剂一次性加入,即传统的“锅煮”式熔融法制备的氨合成催化剂具有最佳的活性;但对于耐热后的催化剂活性,先加碱性助催化剂>助催化剂一次性加入>先加酸性助催化剂,即先加碱性助催化剂制备的催化剂具有较好的耐热性,而先加酸性助催化剂的制备方法是不可取的。尽管先加碱性助催化剂制备的催化剂的耐热性比助催化剂一次性加入略有提高,但由于熔铁催化剂耐热性高、使用寿命长,而且分次加入的制备过程较复杂,所以助催化剂一次性加入,即传统的“锅煮”式熔融法仍然是可行的。
上述试验结果表明,催化剂表面的酸碱性主要是由催化剂前驱体Fe1-xO决定的,制备方法虽然有一定的影响,但基本上改变不了催化剂表面的酸碱性。
作为一种动态原位分析技术,NH3- TPD技术目前被广泛应用于催化剂表面酸性的研究。一般来说,不同的脱附峰代表不同类型的活性中心,脱附峰峰顶温度表征了该中心对NH3的吸附强度,脱附峰的面积代表该中心活性位数量。氨分子在催化剂表面存在2种吸附态,即NH3中N- H键断裂的解离吸附和N原子上的孤对电子与催化剂表面形成的配位吸附。解离吸附为弱吸附,是反应进行的必要条件;配位吸附为强吸附,由于其占据了一定活性位,对反应不利。氨合成催化剂的NH3- TPD图谱如图4所示。
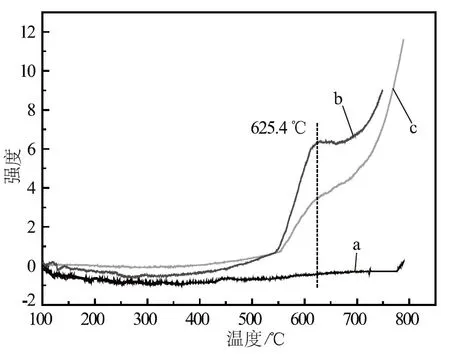
a. 助催化剂一次性加入 b. 分次加入(先加酸性助催化剂) c. 分次加入(先加碱性助催化剂)图4 催化剂的NH3- TPD图谱
由图4可知:在550.0 ℃以下,没有检测到NH3的脱附峰,说明在工业使用温度下,催化剂表面呈碱性。在625.4 ℃,助催化剂分次加入的样品b和样品c均出现了NH3的脱附峰,这属于NH3的强吸附位;而助催化剂一次性加入的样品a在100~800 ℃没有出现NH3的脱附峰,说明改变助催化剂的加入方式改变了高温区的催化剂表面酸性。样品b表面的酸量高于样品c,即样品b表面NH3的强吸附位多于样品c,那么在氨合成反应过程,H2和N2合成的NH3在样品b表面更难脱附。由于被吸附的氨占据了催化剂表面的活性位,故样品b的氨合成活性低于样品c;而样品a不存在NH3的强吸附位,故样品a的氨合成活性最好。
2.3 助催化剂前驱体及其引入方式对催化剂活性的影响
催化剂中的氧化钨除了用于调节酸碱性外,还起着增强催化剂对氮的吸附活化作用。根据研究氨合成反应机理和铁催化剂活性本质得到的结论,氮的吸附离解为速控步骤。但在低温下,催化剂对氮的吸附能力较弱,而对氢较强,故在低温下产生了所谓的氢中毒效应,即H2的强化学吸附现象。为此,必须添加一些对氮有较高亲和力且熔点比铁高的金属或金属氧化物。本发明中的钨有高的亲和力,其在氨合成的氮氢气氛中能被部分还原并与α- Fe形成固溶体,氮以高的键能被吸附在钨上,能明显增强对氮的活化,但这又会使氨合成的第2步——化学吸附氮的加氢变得困难。但在碱金属(钾)存在下,金属和氮键能降低,而且在钨和钾的数量为某确定比例时,其键能可以达到对于过程的2个阶段以可比的速度进行的最佳值。
在熔融法制备催化剂的过程中,存在十分复杂的化学反应,如氧化物前驱体的分解、固溶体的形成等,所以助催化剂的引入方式不同,制备过程发生的化学反应就不同,所得催化剂的性能也不同。因此,试验改变助催化剂的前驱体,如Al2O3和K2O可用KAlO2代替等,这样制备的氨合成催化剂在性能上有一定的差异。试验设计过程主要为:部分助催化剂(Al2O3,K2CO3,CaCO3)在熔融开始前混入熔融炉底部,其他助催化剂在催化剂熔融过程中按CaVO3和CaWO4的混合物、V2O5和WO3的混合物、K2Al2O4,KVO3和K2WO4的混合物3种方式加入。催化剂耐热前后活性随温度变化如图5所示。
a. 助催化剂一次性加入 b. CaVO3和CaWO4后加 c. V2O5和WO3后加 d. K2Al2O4,KVO3和K2WO4后加图5 催化剂耐热前后活性随温度变化
由图5可知:助催化剂分步加入所得催化剂的活性比助催化剂一起加入的高,说明助催化剂分步加入有利于催化剂活性的提高;对于第2步加入助催化剂的方式,助催化剂化合物的引入方式不同,催化剂的活性也有所不同,此种差异在低温(425 ℃和400 ℃)下表现得更为明显;助催化剂钒和钨以其钙盐形式引入比以钾盐形式引入所得催化剂的耐热活性高(耐热前活性差别较小)。
3 结语
(1)助催化剂的添加顺序对催化剂氨合成活性有一定的影响。
(2)助催化剂按其酸碱性顺序加入对催化剂氨合成活性有一定的影响,其中先加碱性助催化剂制备的催化剂的耐热稳定性高于其他方法制备的催化剂。
(3)助催化剂的分步加入有利于催化剂氨合成活性的提高,并且助催化剂钒钨以其钙盐形式引入比以钾盐形式引入所得催化剂的耐热活性高。
[1] 斯拉克A V,詹姆斯 G R.合成氨:第三册[M].北京:化学工业出版社,1980.
[2] FERTIMONT S P A. Process for preparing iron- based catalysts for the synthesis of ammonia and catalysts so obtained:US4789657A[P].1988- 12- 06.
[3] UNIV BOSTON. Partially reduced ferric oxide catalyst for the making of ammonia via the photoassisted reduction of molecular nitrogen and method for the preparation of the catalyst:US4703030A[P].1987- 10- 27.
[4] KLISSURSKI D G, MITOV I G, TOMOV T. A Study Of The Preparation And Properties Of Precipitated Iron Catalysts For Ammonia Synthesis[J]. Studies in Surface Science and Catalysis,1983,16:421- 430.
[5] IMPERIAL CHEMICAL INDUSTRIES PLC. Iron catalyst and method of producing it:US4668658A[P].1987- 05- 26.
[6] MINI RICERCA TECNOLOG. Process for producing the precursor of a precipitated catalyst for the ammonia synthesis:EP0459424A1[P].1991- 12- 04.
[7] ESUEIRAS J, HOMS N, PISCINA P D L, et al. Structure and reactivity of alumina- supported iron catalysts for ammonia synth[J]. Journal of Catalysis,1986(2):264- 276.
[9] SANTOS J, PHILLIPS J, DUMESIC J A. Metal- support interactions between iron and titania for catalysts prepared by thermal decomposition of iron pentacarbonyl and by impregnation[J]. Journal of Catalysis,1983(1):147- 167.
[10] 刘化章.氨合成催化剂——实践与理论[M].北京:化学工业出版社,2007.
[11] LIU Huazhang. Ammonia synthesis catalyst 100 years: Practice, enlightenment and challenge[J]. Chinese Journal of Catalysis,2014(10):1 619- 1 640.
[12] 刘化章,李小年,胡樟能,等.Fe1-xO基氨合成催化剂的制备化学[J].高等学校化学学报,2002(1):87- 91.
[13] 张天明,胡樟能,刘化章.制备条件对FeO基氨合成催化剂性能的影响[J].化学通报(网络版),2003,66:w81.
[14] 刘化章,李小年.Fe1-xO基氨合成催化剂高活性机理初探[J].催化学报,2005(1):79- 86.
[15] 刘化章,李小年.熔铁催化剂活性与其母体铁氧化物形态和组成的关系(I)母体铁氧化物活性次序[J].化工学报,1998(5):534- 541.
[18] 刘化章,李小年,铃木聪雄,等.熔铁催化剂活性与其母体铁氧化物形态和组成的关系(II)表面活性位和氨合成反应速率[J].化工学报,2000(4):462- 467.
[19] 李小年,傅冠平,刘化章,等.助催化剂对Fe1-xO基氨合成催化剂还原性能的影响[J].催化学报,1998(1):24- 28.
[20] 李小年,刘化章,陈诵英.助催化剂对Fe1-xO基氨合成催化剂活性的影响[J].催化学报,1998(3):201- 205.
[21] 李小年,管升,刘化章,等.新体系氨合成催化剂母体中Fe1-xO的歧化及其效应[J].催化学报,1998(4):315- 319.
[22] 李小年,刘化章,许裕生,等.助催化剂对氨合成Fe1-xO基催化剂母体歧化反应的抑制作用的穆斯堡尔谱研究[J].催化学报,1999(1):76- 80.
[23] ALMQUIST J A, CRITTENDEN E D. A Study of Pure- Iron and Promoted- Iron Catalysts for Ammonia Synthesis1[J]. Industrial & Engineering Chemistry,1926(12):1 307- 1 309.
[24] KRABETZ R, PETERS C. The Function of the promotors in the Technical Ammonia Catalyst[J]. Angewandte Chemie International Edition,1965(4):341- 347.
[25] 刘化章,铃木聪雄,大西龙一郎.母体铁氧化物对铁催化剂表面性质和活性的影响[C]∥第九届全国催化学术会议论文集.北京:海潮出版社,1998.
Study of Preparation of Fe1-xO Based Ammonia Synthesis Catalyst by Melting Method Feeding by Stages
HUANG Shiliang, CHENG Tianhong, HAN Wenfeng, LIU Huazhang
(Institute of Industrial Catalysis, Zhejiang University of Technology Zhejiang Hangzhou 310032)
The Fe1-xO based ammonia synthesis catalyst is prepared with melting method, based on properties of oxide cocatalysts including melting point, crystal form and acid- base properties, the effects of feeding method of adding cocatalyst in sequence and by stages during catalyst preparation on catalyst activity and thermal stability are investigated. Results show that adding order of cocatalyst has a certain effect on activity of ammonia synthesis catalyst; adding cocatalysts according to their acid- base property sequence have effects on activity of ammonia synthesis catalyst, the thermal stability of the catalyst, which is prepared by adding alkaline cocatalyst first, is higher than that of other catalysts prepared with other methods; The catalyst which cocatylists vanadium and tungsten are introduced in calcium salt form has higher thermal stability than that catalyst which cocatalyst is introduced in form of potassium salt.
melting method ammonia synthesis Fe1-xO based catalyst preparation method
黄仕良(1988—),男,硕士,从事氨合成催化剂研究。
韩文锋,男,副研究员,从事催化剂工程研究;hanwf@zjut.edu.cn。 刘化章,男,研究员,从事催化剂工程研究;cuihua@zjut.edu.cn。
TQ426.6
A
1006- 7779(2016)06- 0019- 06
2015- 11- 27)
