Molecular cloning and over-expression of a fructosyltransferase from Aspergillusniger QU10
2015-07-19GuoqingZhangJingYangJiajiShiShijunQianandYapengChao
Guoqing Zhang, Jing Yang, Jiaji Shi, Shijun Qian, and Yapeng Chao
Molecular cloning and over-expression of a fructosyltransferase fromQU10
Guoqing Zhang, Jing Yang, Jiaji Shi, Shijun Qian, and Yapeng Chao
State Key Laboratories of Transducer Technology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China
The main commercial production of fructooligosaccharides (FOS) comes from enzymatic transformation using sucrose as substrate by microbial enzyme fructosyltransferase. A fructosyltransferase genomic DNA was isolated fromQU10 by PCR. The nucleotide sequence showed a 1 941 bp size, and has been submitted to GenBank (KF699529). The cDNA of the fructosyltransferase, containing an open reading frame of 1 887 bp, was further cloned by RT-PCR. The fructosyltransferase gene fromwas functionally expressed both inandGS115. The highest activity value for the construction with the α-factor signal peptide reached 431 U/mL after 3 days of incubation. The recombinant enzyme is extensively glycosylated, and the active form is probably represented by a homodimer with an apparent molecular mass of 200 kDa as judged from mobility in seminative PAGE gels. The extracellular recombinant enzyme converted sucrose mostly to FOS, mainly 1-kestose and nystose, liberating glucose. FOS reached a maximal value and represented about 58% of total sugars present in the reaction mixture after 4 h reaction. The results suggest that the availability of recombinantas a new source of a FOS-producing enzyme might result of biotechnology interest for industrial application.
,fructosyltransferase, cloning, expression, fructooligosaccharides
Introduction
Fructooligosaccharides (FOS) as functional food ingredients owing to their prebiotic properties[1-2]are produced either from plant sources or from microorganisms. Fructosyltransferase (EC 2.4.1.9, FTase) and β-fructofuranosidase, also called invertases (EC 3.2.1.26, FFase) are particularly useful for FOS production at industrial level[1,3]. Thus, there is considerable interest in the isolation and characterization of enzymatic activities capable of fructosyl polymerization and also in the genes encoding them[4]. Several microorganisms possessing fructosyl-transferring activity including,,,,sp.,,sp. and yeast were reported[5-8].
Fungi fructosyltransferasecodinggenes have been isolated mainly fromstrains, which genes are more homogeneous with size ranging from 1.6 to 2.2 kb. These genes have been expressed in genetically modified bacteria, yeast, molds and plants, allowing the production of recombinant fructosyltransferases with similar or even better fructosyltransferase activity than observed in native enzyme, and have been used for FOS production[9]. Although successful FOS production using microorganism, especially fungus, has been carried out during the last 20 years, it is still necessary to isolate new genes encoding fructosyltransferases with higher activity.QU10 secretes fructosyltransferase, which has been characterized enzymologically by our laboratory. In this work, we reported the cloning of a fructosyltransferase gene from the.strain and its heterologous expression in(.) and(.). The effect of secretion signal peptide type on the expression level and the use of the recombinant enzyme for FOS production were also investigated.
1 Materials and methods
1.1 Materials
1.1.1 Strains and plasmids
.QU10 (CGMCC3.316) was obtained from the CGMCC (Beijing, China). The.DH5α was used in DNA manipulation. The BL21 and.GS115 (Invitrogen) were used as microbial expression systems. pET-22b (Novagen), pGAPZA and pGAPZαA (Invitrogen) were used as expression vectors.
1.1.2 Culture of.
The fungus was grown on 15% sucrose, 1% yeast extract, 0.5% NaNO3,0.5% K2HPO4, 0.05% MgSO4, 0.05% KCl, 0.2% CMC-Na, adjusted to pH 5.5[10]. The medium was incubated for 72 h at 28−30 °C and 200 r/min.
1.1.3 Genomic DNA and total RNA extraction
Fungal biomass was harvested by filtration through a filter paper and frozen in liquid nitrogen and homogenized by grinding in a mortar. Genomic DNA was extracted using plant genomic DNA kit (Tiangen). Total RNA was isolated using SV total RNA isolated system (Promega). First strand cDNA was synthesized using the GoScript reverse transcript system (Promega) following manufacturer’s instructions.
1.2 Methods
1.2.1 Cloning of.fructosyltransferase gene
PCR primers were designed using the reported sequences of.fructosyltransferase genes. The primer sequences used were sense primer and anti-sense primer[11]or anti-sense primer 2 (GenBank Accession No. DQ233218)[12](Table 1), PCR reactions were carried out using Q5TMHigh-Fidelity DNA polymerase (NEB) and.QU10 genomic DNA or first-strand cDNA as a template, according to the following temperate program: 98 °C for 10 s, 54 °C or 59 °C for 30 s and 72 °C for 80 s for 35 cycles. The amplified DNA fragments were cloned into the pEASY-B vector and sequenced.
1.2.2 Construction of expression plasmids
To construct the expression vectors, fructosyltransferase cDNA was amplified using Q5TMHigh-Fidelity DNA polymerase. Primer P1 and cP2 (Table 1) used for cloning fructosyltransferase cDNA with the native signal sequence, under the following temperature program: 98 °C for 10 s, 59 °C for 30 s and 72 °C for 80 s for 35 cycles. The amplified DNA fragments were digested byRⅠandⅠ, and then cloned into pGAPZA digested by the same restriction enzymes. Primers cP1 and cP2w (Table 1) were used to clone cDNA without the native signal sequence. The PCR products were digested byRⅠandⅠ, and cloned into pGAPZαA digested by the same restriction enzymes. Primers cP11 and cP2w (Table 1) were used to clone cDNA without the native signal sequence. The PCR products were also digested byRⅠandⅠ, and cloned into pET-22b digested by the same restriction enzymes. The presence of the PCR products was verified by restriction enzyme digestion, agarose gel electrophoresis and sequencing.

Table 1 Sequences of primers used in this study
Italicized letters indicate restriction enzyme sites.
1.2.3 Heterologous expression of.fructosyltransferase
To ascertain whether the presumption of the open reading frame of thegene is correct, we tried to express it in.and...BL21 was transformed with pET-22b/cFTSW by heat shock. The engineering bacteria were grown in LB medium until mid-log phase before adding 0.01 mmol/L IPTG. Then FTase expression was continued at 25 °C. Samples were taken at intervals for analysis of biomass yield (600) and fructosyltransferase activity.
.GS115 was transformed with pGAPZA/cFTS and pGAPZαA/cFTSW by electroporation. The plasmids were linearized at theHⅠsite prior to transformation for efficient integration into the.genome. Transformants were selected for Zeocin (Invitrogen) resistance on YPDS (yeast extract-peptone-dextrose, with 1 mol/L sorbitol added) agar supplemented with 100mg/mL Zeocin. Transformants were transferred by toothpicks into 16-well plates, ensuring that each well containing 1.2 mL YPD liquid. The plates were incubated at 28 °C with 180 r/min for 24 h in a humidity shaker. After centrifugation, the culture supernatants were assayed. Colonies with the higher fructosyltransferase activities were transferred by toothpicks into a 250 mL flask with 50 mL YPD liquid media. Samples were taken at intervals for determination of biomass yield (600) and fructosyltransferase activity. The transformant with highest-secreting ability was then screened out.
1.2.4 Sequences analysis
.QU10 fructosyltransferase protein sequence was translated using the DNAMAN, which was also used to align the fructosyltransferase sequences..QU10 protein sequences were analyzed using the ProtParam, NetOGlyc and NetNGlyc tools, available at the Expasy Proteomics Server (http://www.expasy.ch/tools), to identify basic physico-chemical parameters, and number and site of N- and O-glycosilations, respectively.
1.2.5 Enzyme assay
The enzymatic analysis was performed as described by Wang and Zhou[10]. One unit of the transfructosylating activity was defined as 1 µmol of fructose transferred per minute[13].
1.2.6 Seminative polyacrylamide gels analysis of the expressed activity
Seminative PAGE gels were prepared according to Laemmli[14]containing 0.1% SDS, but samples were loaded in a buffer containing the same amount of SDS without β-mercaptoethanol and boiling. After electrophoresis, the gel was washed extensively with 50 mmol/L sodium acetate (pH 5.0) containing 0.5% (/) Triton X-100 for 15 min to remove the SDS. Afterwards, the gel was incubated in 25% sucrose and 50 mmol/L sodium acetate (pH 5.0). Incubated time was 30 min at room temperature. Visualization of sucrolytic activity was performed by incubation with 1% (/) in 2,3,5- triphenyltetrazolium-chloride (TTC) in 0.25 mol/L NaOH at 100 °C. Sucrolytic activity resulted in formation of red formazan dye due to the reaction of TTC with reducing sugars. After a few minutes, the TTC solution was discarded, and the staining was stopped with 5% (/) acetic acid[4,15].
1.2.7 FOS production
The heterologous fructosyltransferase was used for batch production of FOS. The assay was carried out using 250 g/L sucrose at 45 °C in 0.1 mol/L sodium acetate buffer, pH 5.0, and 8 U/mL crude enzyme from recombinant.(measured in the standard assay). Aliquots were taken at intervals. The enzyme was inactivated by boiling for 20 min in water. Then the FOS was analyzed quantitatively by HPLC under the following conditions: column, Hypersil- NH2(250 mm×4.6 mm×5 μL); mobile phase(acetonitrile):(filtered distilled water) = 75:25; flow rate: 1 mL/min; column temperature: 30 °C; refractive index detector (Waters 2414) was used.
2 Results
2.1 Cloning of.fructosyltransferase gene
Sense primer and anti-sense primer 2 were designed (see material and methods) in order to amplify the coding sequence from.QU10. The primers were designed in such a way that the entire fructosyltransferase coding sequence would be amplified. The amplified DNA fragment of about 1.9 kb was cloned into the pEASY-B vector and sequenced. The full-length cDNA was amplified by RT-PCR using the sense and antisense primer 2, Q5TMHigh-Fidelity DNA polymerase, and the first strand cDNA as a template. A single band of about 1.9 kb was obtained and sequenced. The fructosyltransferase genomic DNA consisted of 1 941 bp (Fig. 1A), and the nucleotide sequence has been submitted to GenBank. The obtained cDNA contained an open reading frame of 1 887 bp (Fig. 1B). Sequence comparison of the genomic DNA and cDNA of the.QU10 fructosyltransferase showed the presence of a 54 bp intron located between 1 767 and 1 820 bp (Fig. 2), and the splicing site matched the GT-AG rule[16]. One nucleotide in the fructosyltransferase cDNA was not consistent with corresponding nucleotides of exons from the genomic DNA. It was at position 918 according to the genomic DNA sequence; however, the nucleotide discrepancy did not result in a discrepancy in deduced amino acid sequence.
2.2 Characterization of the deduced protein
The cloned cDNA encodes a protein with 628 amino acids with a deduced molecular mass of 68 kDa. Part of the ORF is a putative signal sequence encoding 15 amino acids[17]. Computational analysis showed that the enzyme has a 4.75 isoelectric point (pI) and 12 potential-glycosylation sites (Fig. 2). The active site was located in Asp64 into the HVLPPNGQIGPCL sequence (Fig. 2), according to the consensus pattern of the glycosyl correspond to the catalytic nucleophile, transition state stabilizer and acid-base catalyst, respectively[18].

Fig. 1 Agarose gel analysis of PCR product of fts gene. (A) PCR amplified fragment of the fts gene from A. niger QU10 genomic DNA. (B) PCR amplified fragment of the fts gene from A. nigerQU10 cDNA. 1: standard DNA marker of different molecular weight; 2: PCR amplified fragment.
The fructosyltransferasegene sequence from.QU10 showed a 94% identity with fructosyltransferase genes from other.strains. Comparison of the amino acid sequences of the.QU10 fructosyltransferase with fructosyltransferase sequences available in database has demonstrated that.QU10 fructosyltransferase has 99% homology with.IFO4308 extracellular invertase[19], followed by 98% homology with.CBS513.88 extracellular invertase (NCBI reference sequence: XP_00139209.2), and 92% with.invertase[20].

Fig. 2 Gene and protein sequences of fructosyltransferase from A. nigerQU10. Intron is shown in lower case letters. N-glycosylation potential sites are represented by asterisks. Nucleotide discrepancy between genomic and cDNA is showed in the white box. The signal peptide is dashed-underlined. Active site is shown in the gray box and active site residue is shown with a double-line.
2.3 Expression of fructosyltransferase in.and.
For heterologous expression of fructosyltransferase, the.strain BL21 and the yeast GS115 were used. Both strains completely lack sucrolytic activity and therefore served an ideal expression system. The fructosyltransferase cDNA without native signal sequence was cloned into the vector pET22b. The resultant plasmids were transformed into.BL21. The enzyme activity was determined in the cell lysate and supernatant. The highest intracellullar fructosyltransferase activity reached 1.68 U/mL. But 0.3% of total fructosyltransferase activity was present in the culture medium. Similar result was reported by Trujillo et al[21]. For yeast transformation, the fructosyltransferasecDNA with native signal sequence was cloned into the vector pGAPZA, under the control of the constitutive promoter GAP. Another cDNA without native signal sequence was inserted downstream of the α factor secretion signal sequence of pGAPZαA. The resultant plasmids and the control vectors were digested byH I, and transformed into GS115, respectively. About 2×102colonies were evaluated for fructosyltransferase expression in 16-well plates. In all cases, the enzyme accumulated activity in the supernatant fluids although at different levels. The fructosyltransferase activity for the construction with α factor secretion signal sequence, which reached 431 U/mL after 3 days of incubation, was 6 fold higher than that for the construction with native signal sequence (63 U/mL). Therefore, the α factor secretion signal peptide is useful for expression in.. It is consistant with previous investigation[22]. However, in other study, replacing the α factor secretion signal peptide with the native signal sequence of laccase resulted in 5 fold higher activity[23]. The biomass yield of the construction with α factor secretion signal sequence was lower than that with native signal sequence (Fig. 3).
2.4 Characterization of the recombinant fructosyltransferase
Molecular mass of the.r QU10 fructosyltransferase protein was estimated to be approximately 100 kDa, as judged by SDS-PAGE run under fully denaturing conditions (Fig. 4A). The high apparent molecular mass of the mature fructosyltransferase protein by SDS-PAGE probably results from post-translational modification. Under these conditions, the intensity of the 200 kDa band seen in seminative PAGE was greatly increased (Fig. 4B). It is to assume that the active enzyme is a homodimer. The heterologous fructosyltransferase exhibited a pH optimum at 5.5, and the optimal temperature of the enzyme was 45 °C. The optimal pH and temperature of the recombinant fructosyltransferase was similar to that of the natural enzyme (data not published).

Fig. 3 Comparison of fructosyltransferase expression using the native and the α-factor secretion signal peptide in P.pastoris GS115. (A) With the native secretion signalsequence. (B) With the α-factor secretion signal sequence.
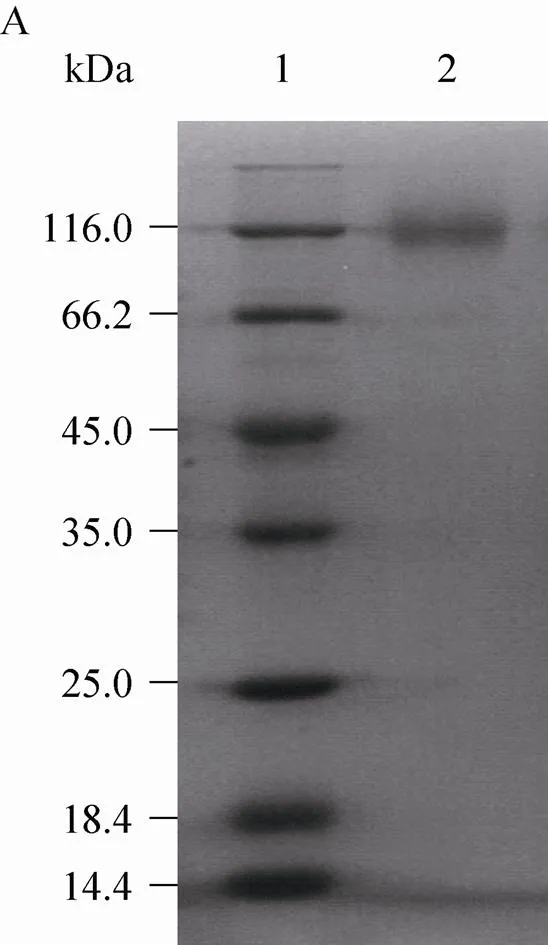
Fig. 4 SDS-PAGE analysis of heterologous fructosyltransferase. (A) Denaturing SDS-PAGE analysis.1: standard protein marker of different molecular weight; 2: culture supernatant of pGAPZαA/cFTSW-GS115. (B) Seminative SDS-PAGE analysis. 1: culture supernatant of pGAPZαA/cFTSW-GS115; 2: standard protein marker of different molecular weight.
The heterologous fructosyltransferase from.was assayed for the production of FOS. A sucrose solution (250 g/L) containing recombinant enzyme (8 U/mL) was incubated at 45 °C in 0.1 mol/L sodium acetate buffer, pH 5.0,and then analysed by HPLC. As shown in Fig. 5, the extracellular recombinant enzyme converted sucrose mostly to FOS, mainly 1-kestose and nystose, liberating glucose. Small amounts (1%) of 1F-fructofuranosylnystose (GF4) were also formed after 8 h (Table 2). The time course of FOS production revealed that more than 84% of the sucrose was consumed by the action of heterologous fructosyltransferase after 4 h of incubation. FOS reached a maximal value and represented about 58% of total sugars present in the reaction mixture. The efficiency of FOS synthesis was about 69%. The trisaccharide kestose reached a maximal value (45% of total sugars present) after 2 h (Table 2).
3 Discussions
In this report, the sense and anti-sense primers were designed. A PCR product of 1.7 kb was amplified with.QU10 genomic DNA as the template. We tried to express the fuctosyltransferase gene with 589 amino acids in.and.. However, we could not express it (data not shown). Based on these results, the extracellular invertase gene sequence from.CBS513.88[12]was selected for the cloning primers design. PCR from genomic and first-strand cDNA samples using the sense primer and anti-sense primer 2 produced a 1.9 kb PCR product (Fig. 1). Sequence comparison of the genomic DNA and cDNA of the fructosyltransferase showed the presence of an intron. As described above, amino acid sequence alignment indicated the fructosyltransferase cDNA product contained an additional C-terminal region consisting of 39 amino acids, which was not present in the amino acid sequence deduced from the PCR product of 1.7 kb. The fructosyltransferasecDNA was successfully expressed and secreted in functional form by.and.. From this result, it was suggested that the extra C-terminal polypeptide was not important for the frutosyl transfer activity but essential for the enzyme production itself. Yanai and Somiari et al get a similar conclusion[11,24].
The sequence comprises an ORF of 1 887 bp encoding a protein of 628 amino acids and thus has a calculated mass of 66.6 kDa, not including a 1 460 Da signal peptide. An increase in mass of about 30 kDa is given that 12 possible N-glycosylation sites could be identified within the sequence of the mature peptide. Due to glycosylation, the apparent molecular mass of the protein by SDS-PAGE was much higher than that calculated for the ORF of(Fig. 4A). Unlike N-glycosylations, the sequences did not present O-glycosylations. Fructosyltransferase sequences in fungi were grouped in two different clades as reported previously[25]. The amino acid sequence of fructosyltransferase from.QU10 belonged to clade VIb. Although a wide variation was observed, a tendency of higher number of N-glycosylations was observed for clade VIb (fungi) sequences.
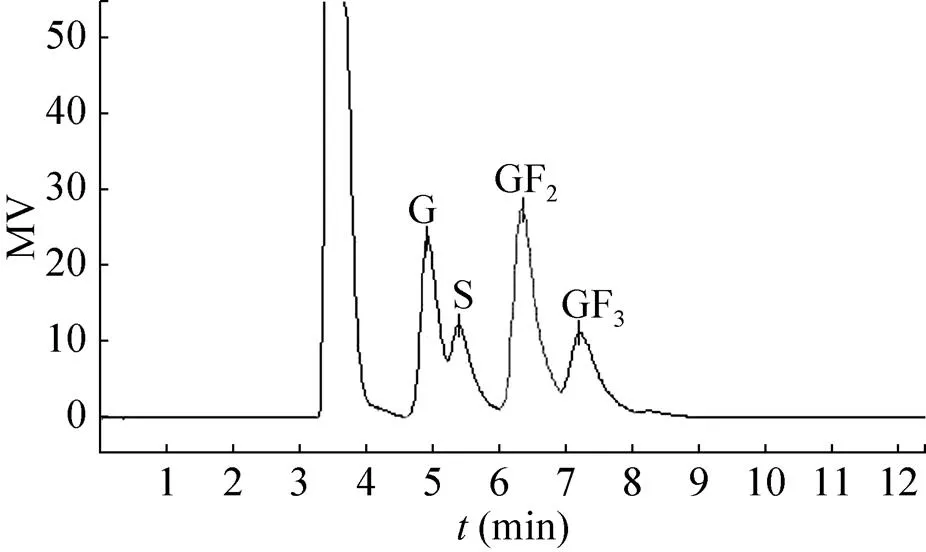
Fig. 5 HPLC chromatogram of oligosaccharides produced from sucrose by heterologous fructosyltranferase. G: glucose; S: sucrose: GF2: kestose; GF3: nystose.
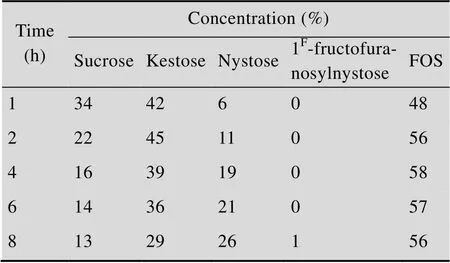
Table 2 Products and substrate concentration variation of heterologous fructosyltransferase during the reaction course
As shown in Fig. 5, FOS reached a maximal value and represented about 58% of total sugars present in the reaction mixture. Therefore, we believe that thegene product has strong fructosyl transfer activity. In the deduced peptide sequence of.QU10 fructosyltransferase, we could recognize that the amino acid sequence between positions 61 and 77 of thegene product is identical to that of thegene product. Thegene and its expression system in this study also will provide a clue as to the mechanism by which thegene product shows strong fructosyl transfer activity[24]. The highest fructosyltransferase activity for the construction with α factor secretion signal sequence reached 431 U/mL after 3 days of incubation. The activity of recombinant fructosyltransferase is approximately 35-fold than that of the natural frutosyltransferase (12.3 U/mL, data not published). Therefore, the recombinant.is more efficient to produce this enzyme. Although the methods of assay are different, it is worth nothing that the activity of recombinant fructosyltransferase is significantly high, compared with other study[21,24].
In conclusion, the recombinant fructosyltransferase from.GS115 has a higher transfructosylating capacity and activity compared with related enzymes, which are advantageous for FOS synthesis. The availability of recombinant.as a new source of a FOS-producing enzyme might result of biotechnology interest for industrial application.
REFERENCES
[1] Maiorano AE, Piccoli RM, da Silva ES, et al. Microbial production of fructosyltransferases for synthesis of pre-biotics. Biotechnol Lett, 2008, 30(11): 1867−1877.
[2] Simmering R, Blaut M. Pro- and prebiotics--the tasty guardian angels. Appl Microbiol Biotechnol, 2001, 55(1): 19−28.
[3] Ghazi I, Fernandez-Arrojo L, Garcia-Arellano H, et al. Purification and kinetic characterization of a fructosyltransferase from. J Biotechnol, 2007, 128(1): 204−211.
[4] Rehm J, Willmitzer L, Heyer AG. Production of 1-kestose in transgenic yeast expressing a fructosyltransferase from. J Bacteriol, 1998, 180(5): 1305−1310.
[5] Yun JW. Fructooligosaccharides-occurrence, preparation and application. Enzyme Microb Technol, 1996, 19: 107–117.
设定测量系统,在相同速度下(设定测量系统运动参数为:stp-20000;spd20),比较3个不同作用点距地高度处的测量值与理论值的关系曲线,如图9所示。
[6] Rodríguez MA, Sánchez OF, Alméciga-Díaz CJ. Gene cloning and enzyme structure modeling of theN74 fructosyltransferase. Mol Biol Rep, 2011, 38(2): 1151−1161.
[7] Sangeetha PT, Ramesh MN, Prapulla SG. Recent trends in the microbial production, analysis and application of fructooligosaccharides. Trends Food Sci Tech, 2005, 16(10): 442−457.
[8] Chuankhayan P, Hsieh CY, Huang YC, et al. Crystal structures offructosyltransferase complex with donor/acceptor substrates reveal complete subsites in the active site for catalysis. J Biol Chem, 2010, 285(30): 23251−23264.
[9] Guio F, Rodriguez MA, Alméciga-Diaz CJ, et al. Recent trends in fructooligosaccharides production. Recent Pat Food Nutr Agric, 2009, 1(3): 221−230.
[10] Wang LM, Zhou HM. Isolation and identification of a novelJN19 producing β-fructofuranosidase and charaterization of the enzyme. J Food Biochem, 2006, 30(6): 641−658.
[11] Somiari R I, Brzeski H, Tate R. Cloning and sequencing of angene coding for b-fructofuranosidase. Biotechnol Lett, 1997, 19(12): 1243−1247.
[12] Yuan XL, Goosen C, Kools H, et al. Database mining and transcriptional analysis of genes encoding inulin-modifying enzymes of. Microbiology, 2006, 152(10): 3061−3073.
[13] Chen WC, Liu CH. Production of β-fructofuranosidase by. Enzyme Microb Technol, 1996, 18: 153−160.
[14] Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 1970, 227: 680−685.
[15] Heyer AG, Wendenburg R. Gene cloning and functional characterization by heterologous expression of the fructosyltransferase ofIAM 2544. Appl Environ Microbiol, 2001, 67(1): 363−370.
[16] Ballance DJ. Sequences important for gene expression in filamentous fungi. Yeast, 1986, 2(4): 229−236.
[17] Pel HJ, de Winde JH, Archer DB, et al. Genome sequencing and analysis of the versatile cell factoryCBS 513.88. Nat Biotechnol, 2007, 25(2): 221−231.
[18] Velázquez-Hernández ML, Baizabal-Aguirre VM, Bravo-Patiño A, et al. Microbial fructosyltransferases and the role of fructans. J Appl Microbiol, 2009, 106(6): 1763−1778.
[19] Futagami T, Mori K, Yamashita A, et al. Genome sequence of the white koji moldIFO 4308, used for brewing the Japanese distilled spirit shochu. Eukaryotic Cell, 2011, 10(11): 1586−1587.
[20] Boddy LM, Bergès T, Barreau C, et al. Purification and characterisation of aninvertase and its DNA sequence. Curr Genet, 1993, 24(1/2): 60−66.
[21] Trujillo LE, Arrieta JG, Dafhnis F, et al. Fructo-oligosaccharides production by thelevansucrase expressed in the methylotrophic yeast. Enzyme Microb Technol, 2001, 28(2): 139−144.
[22] Cregg JM, Vedvick TS, Raschke WC. Recent advances in the expression of foreign genes in. Biotechnology, 1993, 11: 905−909.
[23] Liu W, Chao Y, Liu S, et al. Molecular cloning and characterization of a laccase gene from the basidiomyceteand expression in. Appl Microbiol Biotechnol, 2003, 63: 174–181.
[24] Yanai K, Nakane A, Kawate A, et al. Molecular cloning and characterization of the fructooligosaccharide- producing β-fructofuranosidase gene fromATCC 20611. Biosci Biotechnol Biochem, 2001, 65(4): 766−773.
[25] Alméciga-Díaz CJ, Gutierrez ÁM, Bahamon I, et al. Computational analysis of the fructosyltransferase enzymes in plants, fungi and bacteria. Gene, 2011, 484(1/2): 26−34.
(本文责编 郝丽芳)
黑曲霉QU10果糖基转移酶的克隆表达
张国青,杨敬,石家骥,钱世钧,钞亚鹏
中国科学院微生物研究所传感技术联合国家重点实验室,北京 100101

张国青, 杨敬, 石家骥, 等. 黑曲霉QU10果糖基转移酶的克隆表达. 生物工程学报, 2015, 31(4): 512-522.Zhang GQ, Yang J, Shi JJ, et al. Molecular cloning and over-expression of a fructosyltransferase from Aspergillusniger QU10. Chin J Biotech, 2015, 31(4): 512-522.
微生物果糖基转移酶能够以蔗糖为底物产生低聚果糖。为获得更多新酶资源,通过PCR法成功地克隆出黑曲霉QU10的果糖基转移酶基因(GenBank Accession No. KF699529),基因片段长度为1 941 bp,包含一个54 bp的内含子。进一步利用RT-PCR法克隆了果糖基转移酶的cDNA,其编码628个氨基酸。将所得片段定向克隆到pET-22b、pGAPZA及pGAPZαA载体,并转化至大肠杆菌或毕赤酵母中,通过筛选获得果糖基转移酶表达活力高的转化子。利用α信号肽的毕赤酵母转化子获得最高果糖基转移酶胞外酶活力为431 U/mL,是原始菌株酶活力的35倍。此毕赤酵母重组酶为同源二聚体,半天然PAGE表观分子量约200 kDa。以蔗糖为底物,果糖基转移酶在pH 5.0、45 ℃下反应4 h,酶解产物中主要是蔗果三糖和四糖,蔗果寡糖最高可占总质量的58%。结果表明,果糖基转移酶酵母工程菌具有很高的转果糖基的能力,而且表达活力高,具有潜在的工业应用价值。
黑曲霉,果糖基转移酶,克隆,表达,低聚果糖
August 19, 2014; Accepted: September 25, 2014
Yapeng Chao. Tel/Fax: +86-10-64807428; E-mail: chaoyp@sun.im.ac.cn
Supported by: National High Technology Research and Development Program of China (863 Program) (No. 2012AA101807).
国家高技术研究发展计划(863计划) (No. 2012AA101807) 资助。
网络出版时间:2014-10-14
http://www.cnki.net/kcms/doi/10.13345/j.cjb.140418.html
