家蚕蛋白亚细胞定位预测模型的构建及其初步应用
2015-05-09王小飞石卓兴谭淑敏张耀洲陈剑清舒特俊
王小飞,石卓兴,谭淑敏,李 杰,张耀洲,于 威,陈剑清,舒特俊
(浙江理工大学生命科学学院生物化学研究所,杭州 310018)
家蚕蛋白亚细胞定位预测模型的构建及其初步应用
王小飞,石卓兴,谭淑敏,李 杰,张耀洲,于 威,陈剑清,舒特俊
(浙江理工大学生命科学学院生物化学研究所,杭州 310018)
为研究家蚕蛋白及家蚕杆状病毒蛋白的亚细胞定位并提高预测模型的特异性和准确率,构建了家蚕蛋白亚细胞定位预测模型,并将该模型初步应用于家蚕核型多角体病毒(BmNPV) P10蛋白的亚细胞定位预测中。结果表明,总体预测准确率在不分段时为60.6%,分两段、三段和四段时分别为78.9%、78.4%和80.6%;对BmNPV P10蛋白的预测结果为宿主细胞核,通过免疫细胞荧光实验对预测结果进行验证,结果表明预测结果与实际相符。因此,采用分段的方法能够提高预测准确率,且家蚕病毒蛋白可以利用其宿主家蚕蛋白亚细胞定位预测模型进行亚细胞定位。
家蚕; 亚细胞定位预测; 支持向量机; P10蛋白
0 引 言
预测蛋白质在亚细胞水平的定位,既能为基因组注释、蛋白质功能及其与其他分子的相互作用的推断提供线索,又能为药物靶点的设计提供新的思路。传统的生化实验方法难以满足对大量新发现的未知蛋白的亚细胞定位需求。在已知蛋白亚细胞定位的基础上,结合计算机算法,运用计算机技术可以对蛋白质的亚细胞位点进行准确、高效的预测,有些方法的预测结果甚至比高通量的实验方法更加准确[1]。因此,基于计算预测的方法可以作为传统实验方法强有力的补充甚至替代。
蛋白质的亚细胞定位预测包括三个核心过程:数据集的构建、特征提取和算法设计。过去20多年的发展过程中所涌现出的各种方法大多是围绕这三个核心过程而开发的。数据集有的是用已有的公共数据,有的是针对不同研究课题新建的;提取特征信息的方法主要包括基于蛋白序列的统计信息[2]和基于基因组注释的功能信息[3];设计的算法包括最近邻[4]、人工神经网络[5]、支持向量机[6]和随机森林方法[7]等。目前很多预测模型所使用的数据集都是针对某一生物界的蛋白,比如原核生物蛋白数据集、动物蛋白数据集和植物蛋白数据集等,这些数据集虽然数据庞大,但特异性不高。Shen等[8]提出的方法采用了高维的FunD功能域注释信息,一般此类方法提取的信息维数相等于功能域数据库所有序列个数,这就难免造成了维数灾难。为研究家蚕蛋白的亚细胞定位,本文首次构建了针对家蚕蛋白的亚细胞定位预测数据集,并提出了一种基于融合分段氨基酸组分信息(AAC)和氨基酸位置信息(AAP)的特征提取方法,在分四段的情况下维数也只有160维。采用这种方法维数小,训练速度快,预测准确率也比较理想,且使用方便;所构建的模型基于支持向量机(SVM),在构建的家蚕数据集中取得了比较理想的预测准确率,并成功地将该预测模型初步应用于家蚕病毒蛋白的亚细胞定位研究中。
1 材料与方法
1.1 预测模型的构建
1.1.1 数据集
数据集是从SWISS-PROT数据库提取的家蚕蛋白亚细胞定位数据集,总共包含592条家蚕细胞的蛋白质序列,这些序列分别属于四种亚细胞定位区域:细胞质、细胞膜、细胞核和线粒体。在该数据集中,任意两条序列的相似性均小于50%。为方便起见,将这个数据集命名为Bombyx(表1)。

表1 从SWISS-PROT数据库提取的家蚕蛋白亚细胞定位Bombyx数据集
数据集构建策略:Bombyx数据集的蛋白序列来源于2013年9月15日的SWISS-PROT数据库版本,此版本SWISS-PROT数据库共包含540958条蛋白。数据集提取构建步骤如下:
a) 在SWISS-PORT数据库中输入关键字“家蚕(Bombyx)”;
b) 然后选择“高级搜索(Advanced Search)”;
c) 在高级搜索中选择“AND”,Field选“Subcellular Location”,Term框中输入目标亚细胞位置,比如“cytoplasm”,Confidence选“Any”;
d) 然后点击“Add&Search”,得到以上查询的全部结果;
e) 最后在Reduce sequence redundancy to中选择50%,得到家蚕中细胞质(cytoplasm)蛋白冗余度小于50%的集合;
以上操作也可登陆SWISS-PROT数据库后执行如下查询代码实现:“uniprot: (Bombyx AND annotation: (type:location “cytoplasm”)) identity: 0.5”。
同样步骤可得到其他三个细胞位置的数据,(Term框分别为“membrane”、“nucleus”和“mitochondrion”),最后将检索到的全部数据合并,构成家蚕蛋白数据集。
1.1.2 特征提取
开发蛋白质亚细胞定位的预测方法,第一个要面对的问题就是如何来表示一个蛋白样品,通常会使用两种表示方法:顺序表示和非顺序表示。典型的顺序表示方法就是一个蛋白质的整个氨基酸序列,其中包含了该蛋白几乎所有的信息。而非顺序表示方法采用一个不连续的数据集来表示一个蛋白质,因此又称之为离散表示法,主要利用氨基酸组分、伪氨基酸组分、结构域、基因组注释等方法进行表示[9]。本文主要采用基于20种氨基酸的组分信息和位置信息的方法来提取蛋白质的特征信息。
a) 氨基酸组分信息(AAC)方法
氨基酸组分信息(amino acid composition,AAC)方法[10]是一种基本的蛋白质序列编码方法,它不考虑蛋白质氨基酸残基的顺序信息,仅对20种氨基酸在蛋白质序列中出现的频率进行简单地表示。AAC方法使用20维欧式空间的一个点来表示一个蛋白质序列,用向量表示为:
VAAC(S)=(v1,v2,v3,…,v20)T。
设序列S是由L个氨基酸残基组成的序列,其中,vi=hi/L,hi为第i种氨基酸在序列S中出现的次数(i=1,2,…,20)。虽然AAC计算比较方便,但它具有一个比较大的缺陷,就是它没有考虑一个蛋白质序列的顺序信息,需要使用其他方法来弥补这一缺陷。
b) 氨基酸位置信息(AAP)方法
提取一个蛋白质序列20种氨基酸的组分信息(AAC)的编码方法虽然简单、有效,但同时也会丢失序列中包含的大量顺序信息,为了弥补AAC方法的这一缺陷,本文在AAC方法的基础上又采用了氨基酸位置信息(amino acid position,AAP)[11]的方法来进一步提取蛋白质序列中20种氨基酸的序列信息,与AAC方法类似,AAP方法同样是将一个蛋白质的序列映射到20维欧式空间的一个点,用向量表示为:
VAAP(S)=(v1,v2,v3,…,v20)T。
设序列S是由L个氨基酸残基组成的序列,其中,vi=ri/L,ri为第i种氨基酸在序列S中出现的间隔数之和(i=1,2,…,20),所以vi是表示第i种氨基酸在序列S中出现的间隔系数。
ri的计算下面将用一个例子说明:假如氨基酸A在序列S中出现了5次,分别出现在序列的第3、10、17、28、33个氨基酸位置上,记为PA(S)=(3,10,17,28,33)。
进一步计算氨基酸A在序列S中出现的间隔数,记为GPA(S)=(7,7,11,5),那么ri=(7+7+11+5)/L。
c) 基于分段的AAC和AAP方法
目前利用对蛋白序列进行简单分段的信息提取方法只考虑了蛋白序列局部序列中各个氨基酸所出现的频率,即只是分段的AAC方法,并未考虑局部序列的氨基酸顺序信息以及分段后局部序列对全局序列信息融合的影响。鉴于以上问题,本文采用融合分段氨基酸组成信息及分段氨基酸位置信息的方法对蛋白质的序列信息进行特征提取。
将蛋白序列均分为k个子片段,统计每个子片段的氨基酸组成信息及氨基酸位置信息,再融合成多重信息,能够涵盖片段信息和全局信息。分成k个子片段后,在每个子片段上分别提取20维的AAC和AAP,最终将分别得到k*20维的AAC和AAP,两种信息融合后成为一个k*40维的向量,用这k*40维的向量输入支持向量机SVM(support vector machine)进行学习构建预测模型。基于分段融合的特征提取方法,记为SACP(segmented amino acid composition and position),用向量表示为:
VSACP(S)=(v1,v2,v3,…,vk*40)T(k=1,2,3,…)。
1.1.3 支持向量机
蛋白质的亚细胞定位预测是一个多类的分类问题,本文采用Vapnik提出的支持向量机SVM[12]算法通过组合多个二类分类器来解决这一问题,鉴于训练样本不是很大,本文采用“一对多”的分类策略。首先,SVM把输入向量映射到一个特征空间;然后,SVM在特征空间中寻找最优线性分割来解决二类或者多类问题;最后,为测试样本指定一个预测标签。使用了LIBSVM软件包来实现SVM分类器,选用的核函数是径向基函数(radial basis function,RBF),选择依据是径向基核函数相对于其他核函数在解决非线性问题方面更具优势[13]。
1.2 评价方法和指标
目前对蛋白质亚细胞定位预测结果进行评价的方法主要有以下几种:交叉检验、独立样本检验、刀切法和自相容检验。而刀切法被认为是最严格和最客观的评价方法[14]。刀切法也就是通常所说的留一法,每次取出数据集中的一条蛋白质序列作为测试样本,而将剩余的蛋白质序列作为训练集,依次取出直到所有样本序列都被测试一遍为止。本文选择刀切法对预测结果进行评价。
对SVM分类器标准的性能指标进行了刀切法测试,包括子类准确率Ci,即子类Ci被正确分类的百分数(sub-class accuracy,CA)和总体准确率(overall accuracy,OA),即所有蛋白被正确分类的百分数。子类准确率和总体准确率的公式如下:


其中,TPi代表真阳性的数目,|Ci|代表每个子类Ci所包含蛋白的数目。
1.3 预测模型在BmNPV P10蛋白亚细胞定位预测中的初步应用
1.3.1 BmNPV P10蛋白生物信息学分析
BmNPV p10基因在NCBI(national center for biotechnology information)基因登录号为:L13071.1;利用DNAMAN软件预测家蚕核型多角体病毒(BmNPV)p10基因编码的氨基酸的序列、分子量、等电点、跨膜区、同源性以及进化树;,通过http://www.vivo.colostate.edu/molkit/hydropathy/index.html(Protein Hydropathicity Plots)预测疏水性;通过http://zhanglab.ccmb.med.umich.edu/I-TASSER/(I-TASSER)服务器预测其易溶性。通过I-TASSER(http://zhanglab.ccmb.med.umich.edu/I-TASSER/)服务器预测P10蛋白的高级结构。
1.3.2 BmNPV P10蛋白亚细胞定位预测
上述构建的家蚕蛋白亚细胞定位预测模型也可以用来预测以家蚕为宿主的病毒蛋白的亚细胞定位。因为杆状病毒感染家蚕,其表达的蛋白需要在家蚕细胞内通过家蚕的蛋白识别及转运系统来进行运输,因此,杆状病毒P10蛋白带有家蚕的亚细胞定位信息。使用本研究构建的家蚕蛋白亚细胞定位预测模型对BmNPV P10蛋白的亚细胞定位进行预测。
1.3.3 BmNPV P10蛋白亚细胞定位的免疫细胞方法验证
所用家蚕卵巢上皮细胞(BmN)由本实验保存;野生家蚕杆状病毒由本实验室保存;一抗为自制兔抗P10多克隆抗体,二抗为购买的Alexa Fluor 546 Donkey anti Rabbit抗体。其他试剂参考Abcam的免疫荧光实验方案进行配制。野生家蚕杆状病毒侵染正常BmN细胞,在21 h.p.i.(hours post infection)即极晚期取样进行免疫荧光实验。正常BmN细胞作为对照。
2 结果与讨论
2.1 预测结果及比较
预测结果(表2)表明:不分段时,预测准确率为60.6%,分两段时预测准确率为78.9%,而分四段时预测准确率达到80.6%。说明采用分段的方法能够提高预测准确率,分四段时预测准确率最高。

表2 基于分段统计的Bombyx预测结果
为了验证预测算法的可靠性,将算法应用于由Shen等[8]构建的通用数据集(Virus-mPLoc),该数据集总共包含252条病毒蛋白质序列,这些序列分别属于6种亚细胞定位区域:病毒衣壳、宿主细胞膜、宿主内质网、宿主细胞质、宿主细胞核和宿主细胞外。在该数据集中,任意两条序列的相似性均小于25%。Virus-mPLoc数据集的详细情况如表3所示。

表3 Virus-mPLoc数据集中六种亚细胞位置的蛋白序列个数
从预测结果(表4)可以看到,基于融合分段统计的AAC和AAP信息在Virus-mPLoc数据集中预测准确率与Shen等所提出的方法[2]稍低,在不分段的情况下预测准确率只有37.7%,分两段和分四段时,预测准确率最高,达到42.9%,只比Shen提出的方法低0.8%。由此可以得到结论:采用分段统计的方法能够有效的提高预测准确率。

表4 基于分段统计的Virus-mPLoc预测结果(总体准确率/%)
另外,Shen提出的方法采用了高维的FunD功能域注释信息,一般此类方法提取的信息维数相等于功能域数据库所有序列个数,这就难免造成了维数灾难。相比于笔者提出的基于融合分段AAC和AAP的信息,维数等于k*40,在分四段的情况下维数也只有160维,采用这种方法维数小,训练速度快,预测准确率也比较理想,且使用方便。因此笔者所采用的家蚕蛋白亚细胞定位预测算法具有一定的可靠性。
2.2 预测模型在BmNPV P10蛋白亚细胞定位预测中的初步应用
2.2.1 BmNPV P10蛋白的生物信息学分析
a) BmNPV P10蛋白简介
杆状病毒P10蛋白在核型多角体病毒复制的极晚期大量表达,被感染细胞的细胞质与细胞核中所发现的纤维状结构的主要成分就是P10[15]。P10并非病毒生命周期的必需蛋白,但对于稳定多角体的结构和宿主细胞核的裂解具有重要作用。以往对P10蛋白的研究多是在AcMNPV中,本研究构建的预测模型初步应用于家蚕核型多角体病毒(BmNPV)中的P10蛋白。
b) BmNPV p10基因及P10蛋白的保守结构域及同源性分析
在BmNPV病毒中,编码P10蛋白的基因含有1个213 bp的ORF框,编码70个氨基酸残基的蛋白(图1A),预测其分子量约为7.5 kDa,理论等电点为3.79,无跨膜区,属于NPV_P10超家族(图1D)。通过对杆状病毒P10同源蛋白序列的比对,可以看出,P10蛋白在进化过程中始终保持着十分相似的结构组织形式(图1C),而与BmNPV的P10亲缘关系最近的是AcMNPV的P10蛋白(图1B),两者序列相似度为89%。
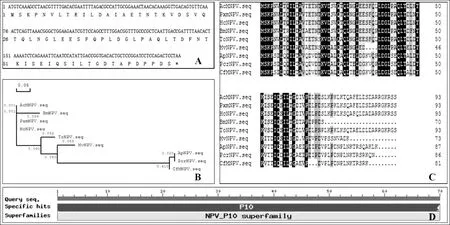
图1 BmNPV p10基因序列及其编码氨基酸序列分析注:A.BmNPV p10基因ORF及其编码氨基酸序列(*代表终止子);B.P10蛋白的进化树,自展值设为1000,图中红色数字代表分支长度;C.杆状病毒P10同源蛋白的序列比对,高亮区代表P10的同源序列;D.BmNPV P10蛋白的保守结构域。
c) BmNPV P10蛋白的水溶性分析
预测结果(图2)可知,该蛋白大部分区域的疏水性预测值都小于0,预测该蛋白属于亲水性蛋白;另外,该蛋白大部分的残基都是暴露在溶剂中,预测该蛋白属于易溶性蛋白。

图2 BmNPV P10蛋白的水溶性分析注:A.BmNPV P10蛋白的疏水性分析,Kyte-Doolittle值大于0代表疏水; B.BmNPV P10蛋白的易溶性预测。
d) BmNPV P10蛋白的高级结构预测
预测结果(图3)表明,BmNPV P10蛋白含有三个α-helix,其蛋白单体的三级结构是由两股反向α螺旋组成的卷曲螺旋,呈杆状,结构和功能类似于FALPE (filament-associated late protein of entomopoxviruses)[16]。

图3 BmNPV P10蛋白的等级结构预测注:(a) 图中C代表Coil(无规卷曲),H代表Helix(螺旋)。
2.2.2 BmNPV P10蛋白的亚细胞定位预测
通过所构建的家蚕蛋白亚细胞定位预测模型,预测BmNPV P10蛋白的亚细胞定位,当不分段时预测结果为P10蛋白定位于宿主细胞的细胞质,分段后的预测结果同为P10蛋白定位于宿主细胞的细胞核,总体准确率最高即分四段时的预测结果为P10蛋白定位于宿主细胞的细胞核。预测结果拟通过细胞的免疫荧光染色实验来验证。
2.2.3 免疫细胞荧光实验验证
从免疫荧光实验结果(图4)可以看出:BmN家蚕细胞被BmNPV病毒感染后,能够在其细胞核内检测到P10蛋白,这一结果与模型的预测结果一致;同时,也能够在其细胞质内检测到P10蛋白,说明P10是一种具有多个亚细胞定位的蛋白。从对P10进行亚细胞定位预测的结果来看,在分段的情形下,预测结果都在宿主细胞核,而在不分段的时候预测结果在细胞质,这两种情况都符合实验情况。但是,细胞质在Bombyx中属于小样本,参考SVM分类器大样本优势的特性,此预测结果比较奇特,有待于进一步研究。
3 讨 论
中国是世界蚕桑最大生产国,蚕桑业在我国农业产业中占有较大比重,对家蚕尤其是家蚕杆状病毒的研究有利于蚕桑业的进一步发展,对病毒蛋白在宿主细胞亚细胞定位的研究有利于进一步研究病毒蛋白的功能和应用价值。本文针对家蚕蛋白特别构建了家蚕蛋白数据集,采用融合分段氨基酸组分信息和氨基酸位置信息的特征提取方法,基于支持向量机算法,构建亚细胞定位的预测模型,取得了比较理想的预测准确率,且准确率随着分段数的增加而提高,最高为分四段时80.6%。该模型还适用于以家蚕为宿主的病毒蛋白在宿主细胞中的定位,将其初步应用于BmNPV P10蛋白的亚细胞定位预测中,免疫荧光实验结果表明预测结果可信,说明此模型有希望开发成为家蚕蛋白质亚细胞定位预测的实用模型,为下一步解决多位点预测问题提供参考。
[1] Rey S,Gardy J L,Brinkman F S L.Assessing the precision of high-throughput computational and laboratory approaches for the genome-wide identification of protein subcellular localization in bacteria[J].BMC Genomics,2005,6(1): 162.
[2] Garg A,Raghava G P S.ESLpred2: improved method for predicting subcellular localization of eukaryotic proteins[J].BMC Bioinformatics,2008,9(1): 503.
[3] Brady S,Shatkay H.EpiLoc: a(working) text-based system for predicting protein subcellular location[C]//Pacific Symposium on Biocomputing.2008,13: 604-615.
[4] Cai Y D,Chou K C.Nearest neighbour algorithm for predicting protein subcellular location by combining functional domain composition and pseudo-amino acid composition[J].Biochemical and Biophysical Research Communications,2003,305(2): 407-411.
[5] Tejedor-Estrada R,Nonell S,Teixido J,et al.An artificial neural network model for predicting the subcellular localization of photosensitisers for photodynamic therapy of solid tumours[J].Current Medicinal Chemistry,2012,19(15): 2472-2482.
[6] Xie D,Li A,Wang M,et al.LOCSVMPSI: a web server for subcellular localization of eukaryotic proteins using SVM and profile of PSI-BLAST[J].Nucleic Acids Research,2005,33(S2): W105-W110.
[7] Li Z C,Lai Y H,Chen L L,et al.Identifying subcellular localizations of mammalian protein complexes based on graph theory with a random forest algorithm[J].Molecular Bio Systems,2013,9(4): 658-667.
[8] Shen H B,Chou K C.Virus-mPLoc: a fusion classifier for viral protein subcellular location prediction by incorporating multiple sites[J].Journal of Biomolecular Structure and Dynamics,2010,28(2): 175-186.
[9] Chou K C,Shen H B.Recent progress in protein subcellular location prediction[J].Analytical Biochemistry,2007,370(1): 1-16.
[10] Cedano J,Aloy P,Perez-Pons J A,et al.Relation between amino acid composition and cellular location of proteins[J].Journal of Molecular Biology,1997,266(3): 594-600.
[11] Dai Q,Wu L,Li L.Improving protein structural class prediction using novel combined sequence information and predicted secondary structural features[J].Journal of Computational Chemistry,2011,32(16): 3393-3398.
[12] Hor C Y,Yang C B,Yang Z J,et al.Prediction of protein essentiality by the support vector machine with statistical tests[C]//Machine Learning and Applications(ICMLA),2012 11th International Conference on.IEEE,2012: 96-101.
[13] Buhmann M D.Radial Basis Functions: Theory and Implementations[M].Cambridge: Cambridge University Press,2003.
[14] Chou K C,Zhang C T.Prediction of protein structural classes[J].Critical Reviews in Biochemistry and Molecular Biology,1995,30(4): 275-349.
[15] Carpentier D C J,Griffiths C M,King L A.The baculovirus P10 protein of autographa californica nucleopolyhedrovirus forms two distinct cytoskeletal-like structures and associates with polyhedral occlusion bodies during infection[J].Virology,2008,371(2): 278-291.
[16] Alaoui-Ismaili M H,Richardson C D.Insect virus proteins(FALPE and p10) self-associate to form filaments in infected cells[J].Journal of Virology,1998,72(3): 2213-2223.
(责任编辑:许惠儿)
Modeling and Preliminary Application of Sub-cellular Localization Prediction Model for Proteins of Bombyx Mori
WANGXiao-fei,SHIZhuo-xing,TANShu-min,LIJie,ZHANGYao-zhou,YUWei,CHENJian-qing,SHUTe-jun
(Institute of Biochemistry,School of Life Science,Zhejiang Sci-Tech University,Hangzhou 310018,China)
In order to investigate the sub-cellular localization of proteins from Bombyx mori and Bmobyx mori nuclear polyhedrosis virus and improve the specificity and accuracy rate of prediction model,the authors developed a prediction model for sub-cellular localization of proteins in Bombyx mori and preliminarily applied it to predict Bmobyx mori nuclear polyhedrosis virus protein P10.The results indicate that the overall accuracy rate of the prediction model is 60.6% when the protein sequence is not segmented.When the protein sequence is divided into two,three and four segments,the overall accuracy rates are 78.9%,78.4% and 80.6% respectively.The prediction result of BmNPV P10 protein is nucleus of its host.The prediction result was verified through immune cell fluorescence experiment.The results show the prediction result conforms to the actual condition.Therefore,segmentation method can improve prediction accurate.In addition,sub-cellular localization can be made for proteins from Bmobyx mori by utilization of its host Bmobyx mori sub-cellular localization prediction model.
bombyx mori; sub-cellular localization prediction; support vector machine; protein P10
1673-3851 (2015) 02-0238-06
2014-03-12
国家高技术发展计划“863”项目(2011AA100603)
王小飞(1987-),男,山东枣庄人,硕士研究生,主要从事生物反应器与蛋白组学方面的研究。
舒特俊,E-mail:peter-shu@126.com
TP399
A
