柱后衍生化高效液相色谱法测定门冬氨酸鸟氨酸颗粒剂的有关物质及含量*
2011-06-21覃婷婷韩晓捷黄哲甦
覃婷婷,韩晓捷,黄哲甦
(天津市药品检验所,天津 300070)
门冬氨酸鸟氨酸颗粒在临床上适用于急、慢性肝病包括肝硬化、脂肪肝、各型病毒性和中毒性肝炎以及高血氨症,特别是因肝脏疾病所引起的中枢神经系统症状的改善及肝性脑病的救治[1]。门冬氨酸鸟氨酸本身紫外吸收较弱,其含量通常采用氨基柱HPLC法通过末端吸收测定[2],或是采用柱前衍生HPLC法[3]测定,这两种方法对于制剂中有关物质的检查均不太适合,前者灵敏度较差,后者衍生试剂干扰较大,且衍生后产物不稳定,影响准确性。《德国药典》2008年版对收载的门冬氨酸鸟氨酸原料规定用TLC法检查有关物质,但是该方法灵敏度不高,无法准确定量。鉴于此,本文尝试建立了柱后衍生的方法测定门冬氨酸鸟氨酸的有关物质,一方面将提高响应值,另一方面,由于柱后衍生反应时间较短,减少了过多杂质的生成,且消除了衍生试剂的干扰,故而具有方便、快速、灵敏度高、重现性好等优点。
1 仪器与试药
LC-2010AHT系列液相色谱仪(日本Shimadzu公司),PINNACLE PCX柱后衍生仪(Pickering公司);门冬氨酸鸟氨酸颗粒(国外某企业,批号005271、612111、709811);对照品:门冬氨酸、盐酸鸟氨酸、盐酸鸟氨酸内酰胺、赖氨酸和Ω-二鸟氨酸(国外某企业,批号分别为BCBC7841、035K05791、ED 12/07、1352773、ED 05/04);辛烷磺酸钠为HPLC专用试剂(山东禹王);乙腈为色谱纯(天津康科得);邻苯二甲醛(J&K公司);巯基丙酸(Alfa Aesar);其余试剂为分析纯。
2 方法与结果
2.1有关物质检查方法
2.1.1色谱条件 色谱柱:Synergi Hydro RP(150 mm×3 mm, 4 μm);流动相A:取1.08 g辛烷磺酸钠溶于1 000 ml水中,加1.6 ml高氯酸;流动相B:取1.08 g辛烷磺酸钠溶于500 ml水中,加500 ml乙腈混匀,再加1.6 ml高氯酸,采用梯度程序洗脱,见表1;流速1.0 ml/min,检测波长336 nm,柱温35 ℃,进样量20 μl。衍生条件温度80 ℃,衍生试剂流速0.6 ml/min。
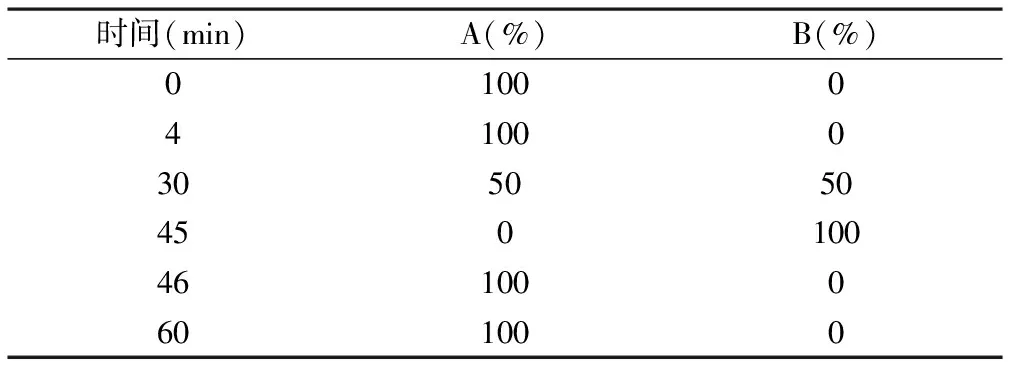
表1 有关物质程序洗脱条件
2.1.2溶液的制备
2.1.2.1供试品溶液的制备 取本品适量,加0.1 mol/L盐酸溶液制成每1 ml中含门冬氨酸鸟氨酸1.5 mg的溶液,作为供试品溶液。
2.1.2.2对照品溶液的制备 分别称取门冬氨酸和盐酸鸟氨酸对照品适量,精密称定,加0.1 mol/L盐酸溶液制成每1 ml中含门冬氨酸和鸟氨酸7.5 μg的溶液,摇匀,作为对照品溶液。
2.1.2.3系统适用性溶液的制备 分别称取门冬氨酸、盐酸鸟氨酸、盐酸鸟氨酸内酰胺、赖氨酸和Ω-二鸟氨酸对照品适量,精密称定,加0.1 mol/L盐酸溶液制成每1 ml中分别含门冬氨酸和鸟氨酸0.75 mg,含鸟氨酰内酰胺、赖氨酸和Ω-二鸟氨酸7.5 μg的溶液,摇匀,作为系统适用性溶液。
2.1.2.4阴性对照溶液的制备 按处方制备不含门冬氨酸鸟氨酸的阴性样品,并按供试品溶液的制法制成阴性对照溶液。
2.1.3系统适用性试验 取系统适用性溶液,进样20 μl,记录色谱图。结果表明,在该色谱条件下门冬氨酸、鸟氨酸与3种杂质分离度均大于1.5,阴性对照在主成分和杂质相应的保留时间处无干扰。各峰的分离度和保留时间见表2,典型分离图见图1。
2.1.4线性考查 精密称取门冬氨酸和盐酸鸟氨酸对照品适量,用0.1 mol/L盐酸溶液配成相当于有关物质检查用对照品溶液的50%、80%、100%、120%和150%浓度的5种溶液,各取20 μl分别注入液相色谱仪,以浓度为横坐标,峰面积为纵坐标得到线性方程。门冬氨酸回归方程为:Y= 42 005X-22 901(r=0.999 6),鸟氨酸回归方程为:Y= 78 041X-38 513(r= 0.999 8)。结果表明,在3.75~11.25 μg/ml范围内门冬氨酸与鸟氨酸溶液浓度均与峰面积呈良好线性关系。
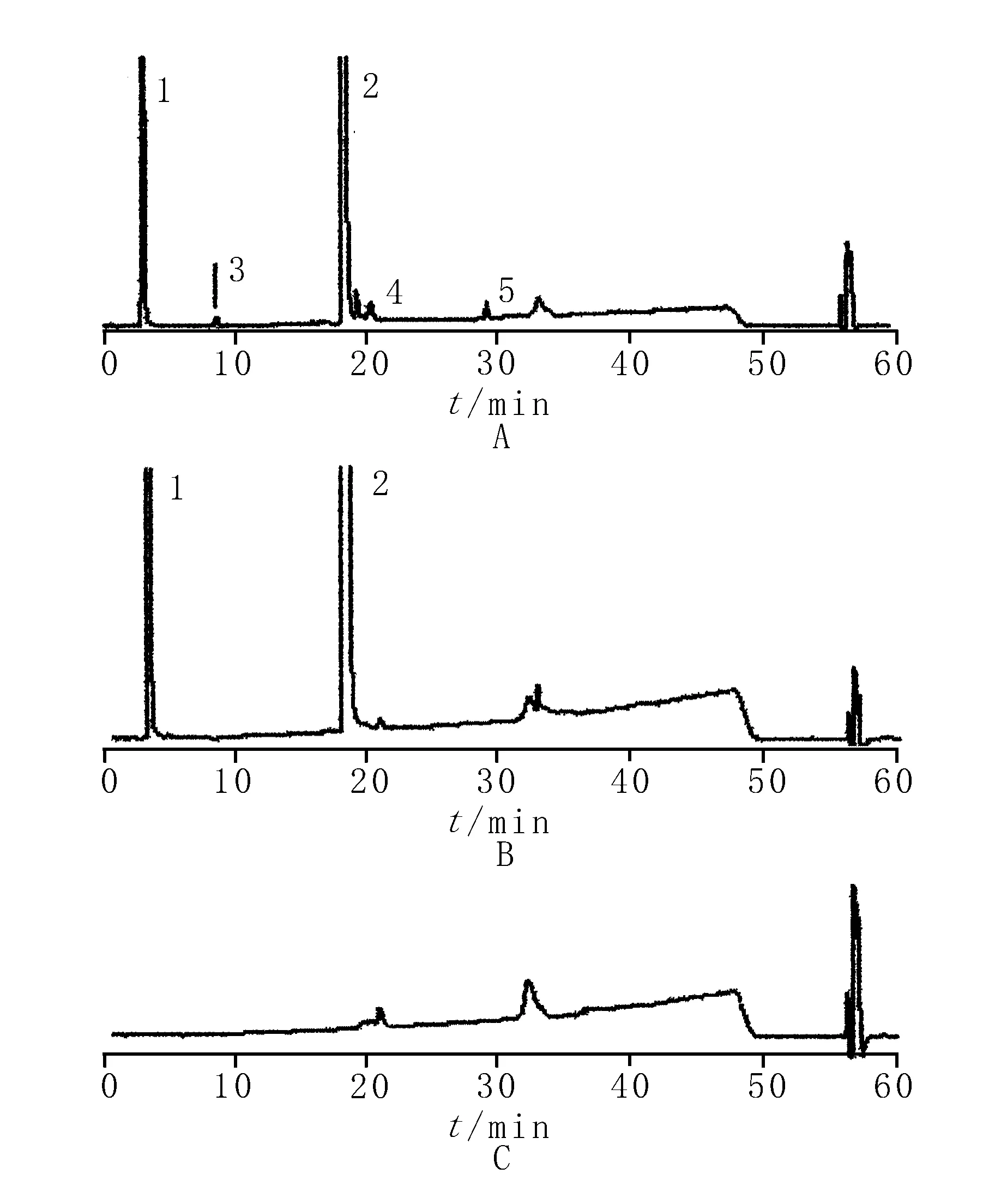
1.门冬氨酸 2.鸟氨酸 3.鸟氨酸内酰胺 4.赖氨酸 5.Ω-二鸟氨酸
2.1.5校正因子的测定 精密称取盐酸鸟氨酸和盐酸鸟氨酸内酰胺对照品适量,用0.1 mol/L盐酸溶液配成每1 ml各分别含4、6、8、10和12 μg的5种溶液,取20 μl分别注入液相色谱仪,以浓度为横坐标,峰面积为纵坐标得到线性方程,以鸟氨酸与鸟氨酸内酰胺的斜率之比作为鸟氨酸内酰胺杂质的校正因子,测得结果为1.851 9。
2.1.6检出限 取“2.1.4”项下的100%浓度溶液,用0.1 mol/L盐酸溶液稀释制备不同浓度的溶液,在选定的色谱条件下,按信噪比为3对最低检测量进行测定。结果表明,门冬氨酸和鸟氨酸的检出限均为5 ng。
2.1.7溶液的稳定性 取配制后的供试品溶液分别于0、2、4和8 h进样,测定峰面积。结果表明,放置4 h后,鸟氨酸内酰胺和未知杂质峰峰面积随放置时间而增高,表明该溶液在4 h内稳定。
2.1.8有关物质测定 取本品3批(批号005271、612111、709811)分别按“2.1.2”项下的方法制备对照品溶液和供试品溶液,进样20 μl,记录色谱图,按外标法加校正因子以峰面积计算,鸟氨酸内酰胺含量按鸟氨酸峰面积计算分别为0.06%、0.08%和0.05%,其他未知杂质的含量,按门冬氨酸峰计算分别为0.09%、0.05%和0。
2.2含量测定方法
2.2.1色谱条件 同“2.1.1”项下色谱条件,程序洗脱条件见表3。
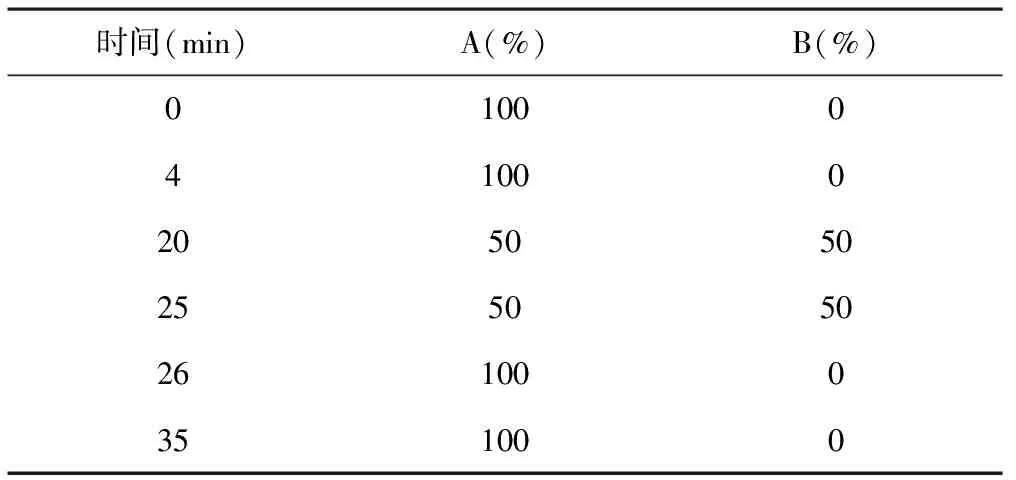
表3 含量测定程序洗脱条件
2.2.2溶液配制
2.2.2.1对照品溶液 分别称取门冬氨酸和盐酸鸟氨酸对照品适量,精密称定,加0.1 mol/L盐酸溶液制成每1 ml中含门冬氨酸和鸟氨酸0.075 mg的溶液,摇匀,即得。
2.2.2.2供试品溶液 取本品10包,研细后精密称定,加0.1 mol/L盐酸溶液制成每1 ml中含门冬氨酸鸟氨酸0.15 mg的溶液,摇匀,即得。
2.2.2.3阴性对照溶液的制备 按处方制备不含门冬氨酸鸟氨酸的阴性样品,并按供试品溶液的制法制成阴性对照溶液。
2.2.3专属性试验 按“2.2.1”项下色谱条件,分别精密量取对照品溶液、供试品溶液和阴性对照溶液各20 μl进样分析。结果表明,对照品溶液中门冬氨酸和鸟氨酸分别在3 min和15.5 min左右出峰,而供试品溶液在相应的位置有相同的色谱峰出现,阴性对照溶液在此处无峰,表明处方中其他成分对主成分含量的测定无干扰,见图2。
2.2.4线性考查 考查了相当于含量测定用对照品溶液的50%~150%范围内的线性关系。门冬氨酸回归方程为:Y= 37 184X- 2 606(r=0.999 8);鸟氨酸回归方程为:Y= 78 062X- 108 174(r=0.999 9)。结果表明,在37.5~112.5 μg/ml范围内门冬氨酸和鸟氨酸溶液浓度与峰面积均呈良好的线性关系。
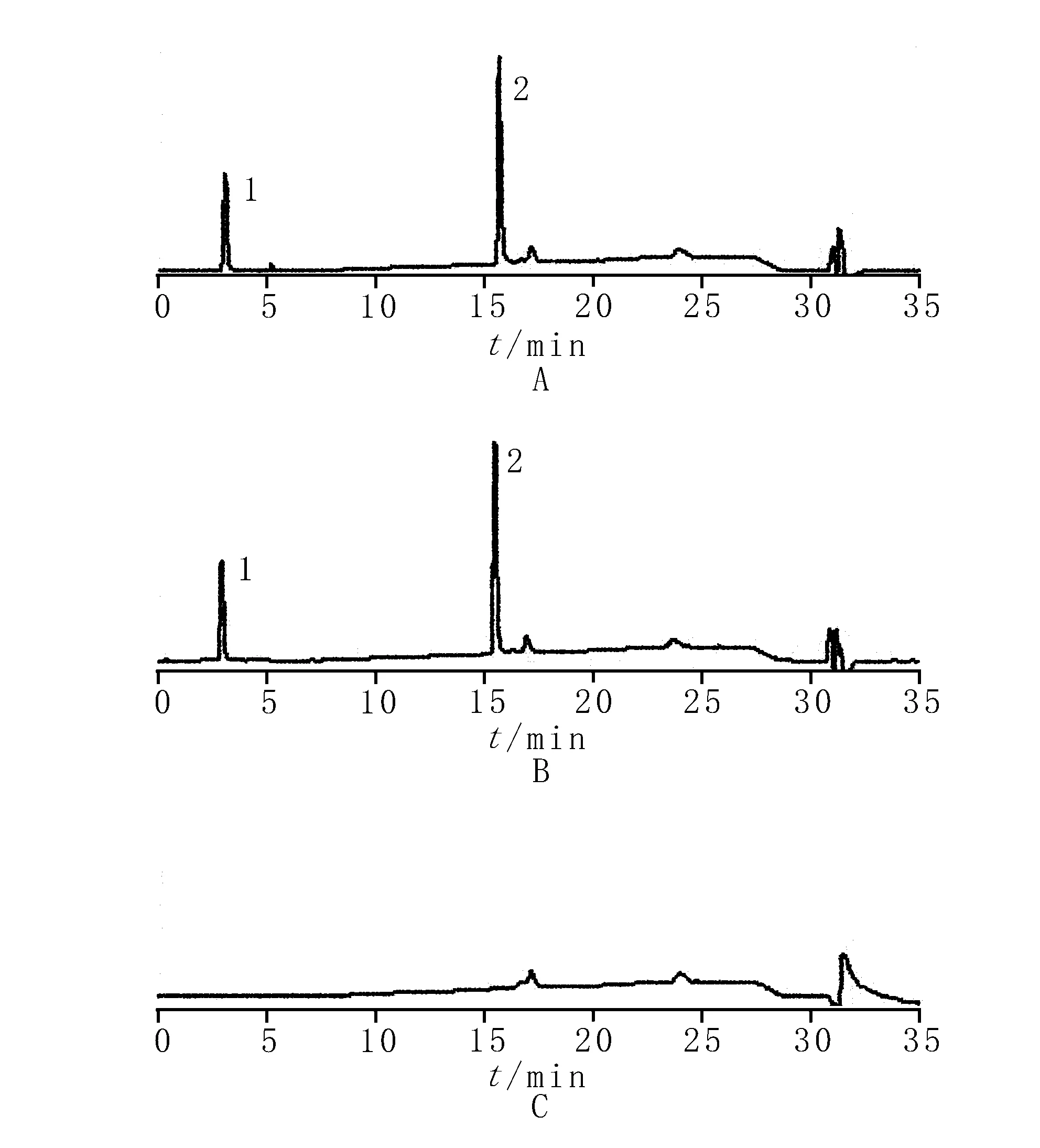
1.门冬氨酸 2.鸟氨酸
2.2.5回收率试验 分别取门冬氨酸对照品60、75和90 mg和盐酸鸟氨酸对照品75、96和115 mg各3份,精密称定,分别置100 ml量瓶中,按处方量分别加入辅料,用0.1 mol/L盐酸溶液溶解并稀释至刻度,再精密量取1 ml至10 ml量瓶中,用0.1 mol/L盐酸溶液溶解并稀释至刻度,摇匀,依法测定并计算门冬氨酸和鸟氨酸的回收率,门冬氨酸回收率(n=9)为101.2%,RSD为0.92%;鸟氨酸回收率(n=9)为101.0%,RSD为0.97%。结果见表4。
2.2.6重复性考查 取同一批样品,同法制备6份供试品溶液,分别测定含量,其平均含量为102.1%,RSD为0.41%,表明方法的重复性良好。
2.2.7供试品溶液的稳定性 取配制后的供试品溶液分别于0、2、4、6、8、10、12和24 h进样,测定峰面积。门冬氨酸峰面积RSD为0.94%(n=8),鸟氨酸峰面积RSD为0.76%(n=8),结果表明,供试品溶液至少在24 h内稳定。
2.2.8含量测定 取“2.2.2”项下的对照品溶液和供试品溶液各20 μl进样,按外标法以峰面积计算样品含量。结果3批样品(批号005271、612111、709811)的含量为102.5%、96.8%和98.9%,均符合规定。

表4 门冬氨酸和鸟氨酸回收率试验结果
3 讨论
3.1色谱柱的选择 在实验过程中曾分别试验过Kromasil C18(150 mm×4.6 mm, 5 μm,)、Agilent SB-C18(150 mm×4.6 mm, 5 μm)等常规规格的色谱柱,但分离效能均没有Synergi Hydro RP(150 mm×3 mm, 4 μm)色谱柱效果好。但是该柱因为内径小,所以柱压高,会缩短色谱柱寿命,并且不易获得,因此在满足分离条件的前提下可以考虑用常规色谱柱代替。
3.2关于有关物质检查方法的建议 考虑到杂质对照品较难获得,采用加校正因子的主成分对照品外标法来测定已知杂质,可减少杂质对照品的使用量,尽量降低分析的成本。
3.3关于含量测定计算的讨论 门冬氨酸鸟氨酸衍生后经色谱柱分离为门冬氨酸和鸟氨酸两个峰,在以前有的计算方法是将二者的峰面积加和计算的,笔者在本实验中是分别计算出门冬氨酸和鸟氨酸的含量后,再分别折合成门冬氨酸鸟氨酸的含量,然后取二者的平均值作为测定的结果。建议折合后的含量应计算相对平均偏差(RAD),将其控制在2%以内,以此保证样品中门冬氨酸与鸟氨酸的摩尔比接近理论值1∶1,使结果更加准确。
本法与薄层色谱法、柱前衍生化HPLC法比较,具有灵敏度高、准确检测制剂中杂质、结果准确等优点,适用于测定门冬氨酸鸟氨酸颗粒剂中的有关物质及含量。
1 蒋音, 陈良, 巫善明. 门冬氨酸鸟氨酸的药理作用与临床应用. 世界感染杂志, 2007, 7(5): 415
2 缪宁梅, 马婕. 高效液相色谱法测定门冬氨酸鸟氨酸注射液的含量. 中国生化药物杂志, 2007, 28(1): 52
3 张莉, 黄哲苏, 李海生. 异硫氰酸苯酯柱前衍生法测定门冬氨酸鸟氨酸注射液含量. 药物分析杂志, 2008, 28(7): 1174
